 Review Article
Review ArticleAbstract
Interstitial lung disease is a rare disorder that has various etiologies and clinical presentations. Some interstitial lung diseases are more common in infancy period, which is not similar to interstitial lung disease in childhood or adult. Every infant presenting with early respiratory distress/failure or chronic respiratory symptoms without reasonable cause together with diffuse pulmonary infiltration should be sought out for interstitial lung diseases after excluding other common conditions of diffuse lung disease. This review focuses on interstitial lung disease in infant or in first year of life in the scope of classification system, systematic approach, investigations, clinical presentations, imaging findings, pathological findings, managements and prognosis. Early recognition of clinical and characteristic of imaging findings of ILD in infants can lead to prompt diagnosis and proper management.
Abbreviations: ILD: Interstitial Lung Disease; chILD: Childhood Interstitial Lung Disease; ACDMPV: Alveolar–Capillary Dysplasia With Misalignment Of Pulmonary Vein; NEHI: Neuroendocrine Cell Hyperplasia Of Infancy; PIG: Pulmonary Interstitial Glycogenosis; SF-B: Surfactant Protein B; SF-C: Surfactant Protein C; ABCA3: Adenosine Triphosphate Binding Cassette Transporter Protein Subfamily A Member 3; PFT: Pulmonary Function Test; FEV1: Forced Expiratory Volume in one Second; FVC: Forced Vital Capacity; CPI: Chronic Pneumonitis of Infancy; DIP:-Desquamative cell Interstitial Pneumonia; NSIP: Nonspecific Interstitial Pneumonia; PAP: Pulmonary Alveolar Proteinosis; TTF-1: Thyroid Transcription Factor
Introduction
“Interstitial lung disease (ILD)” or childhood interstitial lung disease (chILD) is a heterogeneous group of disorders affecting mainly in pulmonary interstitium. Further studies found that many of diseases also have abnormality involve airway and airspace compartment. The term diffuse lung disease may be more accurate [1]. Compare to adult, chILD occurs less frequent. The incidence of chILD varies because of different case definitions and methods of case collection. Incidence studied in Denmark was 10.8 cases/100,000 children/year (1995–2000) and 16.2 cases/100,000 children/year (2001–2005). Incidence studied in German was 0.13 cases/100,000 children and the prevalence rate in United Kingdom and Ireland was 0.36 cases/100,000 children but these maybe underestimate and until now there is no study declare true prevalence or incidence of chILD [2,3].
Diagnostic Criteria and Classification
ILD in adults has been classified base on histopathology [4], but disorders in adults are not similar to children and more different in neonates, thus different approaches in infants are needed. Tachypnea is the most common symptom, which occurs in 75-93% of patients. Other findings are cough, wheeze, crackles, hypoxemia, clubbing of finger and failure to thrive [5-8]. Severe respiratory failure in neonatal period can present in some case. In some infants, the disease is asymptomatic and characterized by occasional findings of diffuse lung disease from CXR [7]. According to American Thoracic Society clinical practice guidelines, ILD in infants will be suspected when an infant (< 2-year-old) has at least three out of four of the following criteria:
a) Respiratory symptoms (e.g., cough, rapid and/or difficult breathing, or exercise intolerance).
b) Respiratory signs (e.g., resting tachypnea, adventitious sounds, retractions, digital clubbing, failure to thrive, or respiratory failure).
c) Hypoxemia.
d) Diffuse abnormalities on CXR or a CT scan.
In addition to above criteria, the common causes of diffuse lung disease which include cystic fibrosis, congenital or acquired immunodeficiency, congenital heart disease, bronchopulmonary dysplasia, pulmonary infection, primary ciliary dyskinesia must be excluded [9].
Classification
Deutsch GH et al. proposed the first classification of diffused lung disease in childhood, later also used by most guidelines [9-11].
Diffuse Developmental Disorders
a) Acinar dysplasia
b) Congenital alveolar dysplasia
c) Alveolar–capillary dysplasia with misalignment of pulmonary vein (ACDMPV)
Growth Abnormalities
a) Pulmonary hypoplasia
b) Chronic neonatal lung disease
• Prematurity-related chronic lung disease (bronchopulmonary dysplasia)
• Acquired chronic lung disease in term infants
c) Structural pulmonary changes with chromosomal abnormalities
• Trisomy 21
• Others
d) Associated with congenital heart disease in chromosomally normal children.
Specific Conditions of Undefined Etiology
a) Neuroendocrine cell hyperplasia of infancy (NEHI)
b) Pulmonary interstitial glycogenosis (PIG)
Surfactant Dysfunction Mutations and Related Disorders
a) SF-B genetic mutations
b) SF-C genetic mutations
c) ABCA3 genetic mutations
d) Others with histology consistent with surfactant dysfunction disorder without a yet recognized genetic disorder
Definition of Abbreviations
SF-B: surfactant protein B, SF-C: surfactant protein C, ABCA3: adenosine triphosphate binding cassette transporter protein subfamily A member 3.
Diagnostic Tools
Echocardiogram
Echocardiogram is useful for excluding congenital heart disease and pulmonary vascular disease that can mimic or coexist with chILD. Furthermore, echocardiogram can evaluate degree of pulmonary hypertension that associated with poor prognosis [12]. So, echocardiogram is strongly recommended in all suspected of chILD [9].
Pulmonary Function Tests (PFT)
PFT in infants may be a challenge task and limited in some centers. Patients need to be sedate and require supplemental oxygen therapy to perform the raised-volume rapid thoracic compression method [13]. Generally, among cases of ILD, pulmonary function abnormalities show restrictive ventilatory pattern with reduce lung compliance, FEV1, FVC and decreased lung volume [14]. In Neuroendicrine cell hyperplasia of infancy (NEHI), PFT is characterized by hyperinflation and airflow obstruction pattern.
Imaging Studies
CXR: CXR is usually the initial imaging study. Most CXR in infants with ILD reveal diffuse opacities related to hypoinflation [15] but rarely provide specific diagnosis. Nevertheless, normal CXR cannot exclude chILD (Figures 1-3).
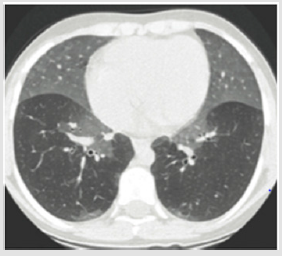
Figure 1: CT imaging if NEHI infant shows ground glass opacification prominent in lingular and middle lobe. This figure was permitted and provided from reference [28].
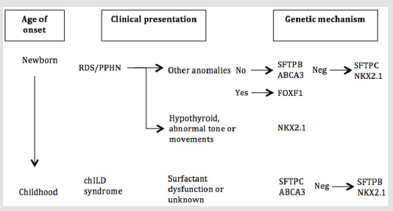
Figure 2: Genetic approach to childhood interstitial lung disease diagnosis. RDS = respiratory distress syndrome; PPHN = persistent pulmonary hypertension of the newborn. This approach was adapted from American Thoracic Society [9].
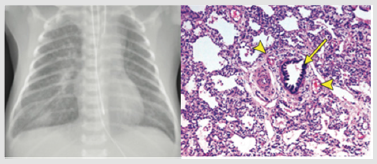
Figure 3: A. The CXR of 5-day old infant presented with respiratory failure shows bilateral reticular infiltration and ground-glass opacification with perihilar bronchial wall thickening, more prominent on the right, and a small right pleural effusion. B. There is a bronchiole (arrow) with an accompanying artery. There is concentric muscular thickening of the arterial media, indicating pulmonary arterial hypertension. At the 4 o’clock and 11 o’clock positions accompany to bronchiole there are two small veins (arrowheads), indicating misalignment of the pulmonary veins. There are also numerous dilated lymphatics, indicating lymphangiectasia, which is also a feature in this condition. This figure was permitted and provided from reference [28].
High-Resolution CT (HRCT): HRCT is the imaging test of choice when ILD is suspected [16]. Nevertheless, HRCT in infants is difficult due to small body size, movement, rapid breathing and inability to hold breath; therefore, it is often necessary to perform this diagnostic assessment under sedation. Using CT scanning protocol of intubation, lung recruitment, and controlled ventilation at centers with pediatric chest CT expertise is recommended [9,17]. Most common HRCT findings in chILD consist of varying combinations of air trapping, ground-glass opacities, consolidations, cysts, nodules and linear/reticular opacities [18]. In some conditions, for example NEHI, HRCT can be used for diagnosis without lung biopsy with 78% sensitivity and 100% specificity (Figure 1) [19].
Magnetic Resonance Imaging (MRI): MRI is inferior to HRCT in resolution, detecting characteristics of chILD diseases and correlating with histological findings. However, techniques of MRI for lung imaging are improving rapidly, comparison of HRCT and MRI could be necessary from time to time [9].
Genetic Testing
Genetic sequencing with whole exomes or whole genomes of the patient and both parents are useful [20]. Several single-gene disorders have been identified that can result in chILD, therefore genetic testing is recommended. The sequence of which genes to analyze depends on age at presentation, mode of inheritance, extrapulmonary manifestations and other factors (Figure 2) [9]. For instance, newborn with severe hypoxemia, respiratory failure and pulmonary hypertension may result from mutations in SP-B, ABCA3 and alveolar–capillary dysplasia with misalignment of the pulmonary veins (ACDMPV). ACDMPV will be highly suspected if patients have extrapulmonary involvement [21].
Bronchoalveolar lavage (BAL)
BAL can aid the diagnosis in some conditions of chILD. There is one study suggests that increase in number of pro-surfactant protein C in BAL may help in diagnosis of SP-C mutations [22]. However, the findings of BAL do not truly reflect the cellular composition of the interstitial space. For this reason, identifying specific chILD from BAL still has limitation. The benefit of bronchoscopy with BAL in patients suspected of chILD is mainly to exclude infection [23].
Surgical Lung Biopsy
Lung biopsy is more likely to determine a specific chILD disease than less invasive procedures [8] and pathologic findings are used for the chILD classification [10]. Video-assisted transthoracic surgery (VATS) biopsy has fewer surgical complications, shorter duration of operation and less pain compare with open thoracotomy, while providing a similar diagnostic yield. This procedure is recommended by American Thoracic Society. For diagnostic accuracy, proper tissue transportation and expertise pediatric lung disease pathologist are essential (Figure 3) [9].
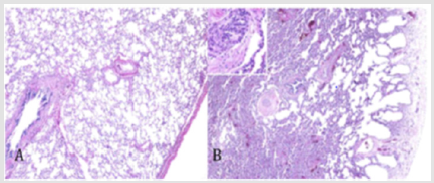
Figure 4: Lung growth abnormalities. Compared with lung from a normal term infant (A, hematoxylin and eosin [H&E], 340) there is reduced alveolarization in a patient with Trisomy 21, characterized by cystic dilatation of subpleural alveoli (B, H&E, 340) This figure was permitted and provided from reference [10].
Childhood Interstitial Lung Disease More Prevalent in Infancy
Diffuse Developmental Disorders
These conditions compose of acinar dysplasia, congenital alveolar dysplasia and alveolar capillary dysplasia with misalignment of pulmonary veins (ACDMPV). The etiology of disorders is believed to be due to aberrations in primary molecular mechanism of lung and/or vascular development [10]. Infants with these disorders usually present as term newborns with rapid, progressive respiratory distress, hypoxemic respiratory failure and pulmonary hypertension without response to medical therapy or extracorporeal membrane oxygenation. Imaging typically shows decrease lung volume with diffuse opacities. In some cases, increase of main pulmonary artery and increase in pulmonary blood flow may be seen if affected infants have concurrent pulmonary hypertension. Unfortunately, most of the patients with these disorders die in the first two months of life [24].
Acinar Dysplasia: Acinar dysplasia is the rarest and the most severe form of diffuse developmental disorders [24]. Some referred to as type 0 congenital pulmonary airway malformation (CPAM). Infants with acinar dysplasia can present at birth and their survival is shortest. This abnormality effect lung maturation at the pseudo glandular to canalicular phase [25]. Therefore, pathologic feature reveals a complete lack of alveolar development and has only airway-like structures [11], which usually found by autopsy.
Congenital Alveolar Dysplasia: The lung development arrests slightly later, at late canalicular to saccular phase. Hence, histology shows acinar formation with simplified lung structure [26]. These infants also present with severe lung disease and pulmonary hypertension with slightly response to supportive treatments [25].
Alveolar Capillary Dysplasia with Misalignment of Pulmonary Veins (ACDMPV): In ACDMPV, a specific stage in lung development is not clearly reminiscent. Clinical presentation can be early within the first few days of birth or delay to a few months of ages [24]. ACDMPV should be suspected in a term infant who has severe pulmonary hypertension, especially in the absence of risk factors such as perinatal depression, meconium aspiration or sepsis [25]. Furthermore, apart from pulmonary manifestation, 50- 70% of ACDMPV can have extrapulmonary anomalies, including gastrointestinal (malrotation, alimentary tract atresias), cardiac (coarctation of aorta, hypoplastic left heart syndrome), absent or hypoplastic gallbladder, and genitourinary anomalies [16]. ACDMPV named from the abnormality of pulmonary pathology in which pulmonary veins run in the same bronchovascular bundle as the pulmonary arteries (the “misaligned veins”) [25]. Other findings are thickening of the alveolar septa from excessive mesenchymal cells and myxoid extracellular matrix, lack close apposition of the capillaries to the alveolar basement membranes, medial hypertrophy of the pulmonary arterioles and prominent diffuse or regional lymphangiectasis [27,28] as show in Figure 3. Because of familial cases are reported, recent studies suggest genetic basis for this disorder. Microdeletion/mutation of the FOXF1 gene which encodes a transcription factor for vascular development have been analyzed in ACDMPV infants [29]. Therefore, genetic testing allows a definitive diagnosis for ACDMPV without performing invasive procedure on unstable child. Nevertheless, genetic testing does not have 100% sensitivity and the absence of mutation cannot exclude diagnosis of ACDMPV because FOXF1 variant deleterious can occur [30].
Growth Abnormalities
Growth abnormalities or alveolar growth disorders are the most common cause (25%) of chILD in infants aged < 2 years [10]. Abnormal pulmonary development is secondary, unlike diffuse developmental disorder that is programed to be abnormal. This category include
1. pulmonary hypoplasia associated with prenatal conditions; restriction of fetal thoracic space, oligohydramnios, skeletal anomalies, neuromuscular problems resulting in decreased or absent fetal breathing movements
2. postnatal conditions; prematurity-related chronic lung disease (bronchopulmonary dysplasia) and acquired chronic lung disease in term infants
3. the structural pulmonary changes seen with a variety of chromosomal abnormalities especially in Trisomy 21
4. Congenital cardiac abnormalities that limiting pulmonary blood supply result in deficient lung growth.
Depending on underlying conditions, affected patients have varying degree of respiratory distress and hypoxemia but generally present in first month of life. Clinical may improve because of new alveoli develop, on the other way, clinical can be worsen in ongoing alveolar injury. As mention above, growth abnormalities may suspect in infants with specific history and associated conditions. Unexplained or severe pulmonary symptoms are the indication for lung biopsy [27]. In bronchopulmonary dysplasia and other conditions, imaging shows hyperexpansion and hyperlucent areas from alveolar enlargement, linear ground glass opacities from fibrotic change and reduction of distal vascularization. In Trisomy 21, airspace are decreased and enlarged prominently in peripheral region, these changes can reveal subpleural lung cyst on HRCT as show in Figure 2 [31]. Histologic features in growth abnormalities are alveolar enlargement and lobular simplification. Since there are multiple associated conditions in this group of ILD, outcomes vary. Overall mortality is 34%, moreover, mortality may be higher in prematurity, severe alveolar growth abnormality and pulmonary hypertension groups [27].
Specific conditions of undefined etiology
Neuroendocrine Cell Hyperplasia of Infancy (NEHI): Neuroendocrine cell hyperplasia of infancy (NEHI) known as persistent tachypnea of infancy, or chronic idiopathic bronchiolitis of infancy is a disorder of unknown etiology. Affected children are typical term infants present with significant tachypnea, retraction, creackle and hypoxemia in first year of life. Respiratory failure is uncommon [10]. HRCT shows air trapping in mosaic attenuation pattern or geographic ground glass opacity involves at least 4 lobes, predominantly in lingular and right middle lobe [32]. These finding are very specific (100%) for NEHI [19], thus patients can be diagnosed without tissue biopsy. If biopsy is performed, histologic findings reveal increase bombesin-immunopositive neuroendocrine cells in bronchioles with nearly normal lung histologic background as show in Figure 5, but this finding also can be found in other disorders [33,34]. Infants with NEHI usually have clinical improvement from time to time, although some may require supplement oxygen until childhood [35]. No deaths, no respiratory failure and no end-stage pulmonary disease were reported directly related to NEHI [24].
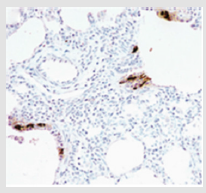
Figure 5: Histologic findings in NEHI infant shows an increase in bombesin-immunopositive cells (brown staining) in the epithelium of the bronchioles. This figure was permitted and provided from reference [28].
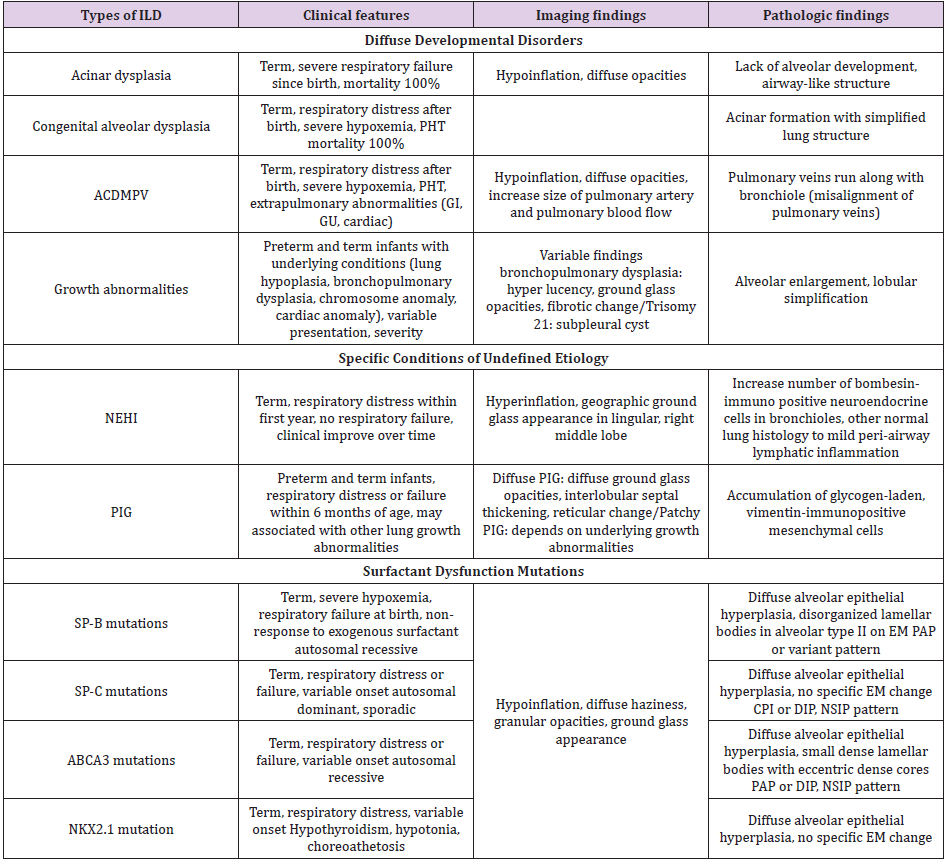
Table 1: Summary of clinical, imaging and pathologic findings of interstitial lung disease more prevalent in infancy.
Note: ACDMPV = alveolar capillary dysplasia with misalignment of pulmonary veins; NEHI, = neuroendocrine cell hyperplasia of infancy; PIG = pulmonary interstitial glycogenosis; PHT = pulmonary hypertension; GI = gastrointestinal; GU = genitourinary; SP-B = surfactant protein B; SP-C = surfactant protein C; ABCA3 = adenosine triphosphate binding cassette transporter protein subfamily A member 3; EM = electron microscopy; PAP = pulmonary alveolar proteinosis; CPI = chronic pneumonitis of infancy; DIP = desquamative cell interstitial pneumonia; NSIP = nonspecific interstitial pneumonia. This summary was adapted from reference 24.
Pulmonary Interstitial Glycogenosis (PIG): PIG previously known as infantile cellular interstitial pneumonitis and histiocytoid pneumonia [36]. This is a rare disease with unknown etiology. PIG usually present with tachypnea, retraction and hypoxia soon after birth or not more than 6 months of age both in term and preterm infants [10]. There are two types of PIG (1) diffuse PIG which occurs as an isolated condition (2) patchy PIG which associated with underlying lung growth disorder [37,38]. On HRCT, infants with diffuse PIG show diffuse ground glass opacities, reticular changes and hyperinflation in subpleural area [27]. In patchy PIG imaging may overlap with other underlying growth abnormalities. Because of non-specific imaging finding unlike NEHI, PIG requires lung biopsy for diagnosis. Characteristic finding is accumulation of monoparticulate glycogen in immature vimentin-immunopositive mesenchymal cells without underlying inflammation or fibrosis [37,38]. Clinical outcome of patchy PIG may be severe due to related conditions and high mortality is reported. On the other hand, diffuse PIG has excellent prognosis (Table 1) [39].
Surfactant Dysfunction Mutations and Related Disorders
Genetic mutations of encoding proteins that involved in surfactant production, function and metabolism lead to surfactant disorders. In this group, inheritance patterns, clinical presentations and age of onset may overlap. Histologic findings can be chronic pneumonitis of infancy (CPI), desquamative cell interstitial pneumonia (DIP), nonspecific interstitial pneumonia (NSIP), or pulmonary alveolar proteinosis (PAP). Therefore, genetic analysis is essential for establishing a specific diagnosis.
SP-B genetic mutations: Surfactant protein B deficiency is an autosomal recessive disorder. 70% of cases of SP-B deficiency are 121ins2 mutation, this result in unstable transcription, absence of pro- and mature SP-B protein [40]. Most infants with this mutation present with rapidly tachypnea, respiratory failure in term newborn and require mechanical ventilation within hours after birth. CXR and HRCT in affected infants show decrease lung volume, diffuse haziness with granular opacity mimic hyaline membrane disease in preterm newborn. But unlike hyaline membrane disease, SP-B deficiency patients are transiently or minimally response to exogenous surfactant and extracorporeal membrane oxygenation [11]. Apart from genetic sequencing, lung biopsyelectron microscopy findings of disorganized lamellar bodies in alveolar type II cells suggest the diagnosis. Histologic findings are characterized by granular eosinophilic accumulation, periodic acid- Schiff positive, lipo proteineous material [39].
SP-C genetic mutations: Surfactant protein C deficiency is an autosomal dominant disorder, but half of cases occur sporadic by spontaneous protein mutation. SP-C mutations lead to decrease endogenous SP-C secretion, aberrant surfactant protein folding and alveolar type II cells apoptosis [41]. Onset of symptoms of SP-C mutations is variable, patients usually present in late infancy to early adulthood and also presentation of patients can vary from mild respiratory distress, interstitial pneumonia to pulmonary fibrosis [42].
ABCA3 genetic mutations: Mutation of adenosine triphosphate binding cassette transporter protein subfamily A member 3 (ABCA3) is an autosomal recessive disorder. Within surfactant dysfunction mutations group, this is the most common genetic causes [43]. Lack of ABCA3 function leads to reduction of mature SP-C and affects the process of pro SP-B to mature SP-B. In severe mutations, clinical develop shortly after birth, whereas in milder mutations, clinical can present in childhood [44]. Ultrastructure abnormalities in alveolar type II cell aid diagnosis with small dense lamellar bodies with eccentrically placed dense cores as show in Figure 6 [24]. HRCT findings in both SP-C and ABCA3 mutations include ground glass opacity, patchy consolidation, septal thickening or small cystic lucency but none are specific and also histological findings [11].
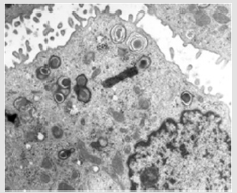
Figure 6: Electron micrograph of ABCA3 mutations patient demonstrates abnormal lamellar bodies with dense inclusions. This figure was permitted and provided from reference [10].
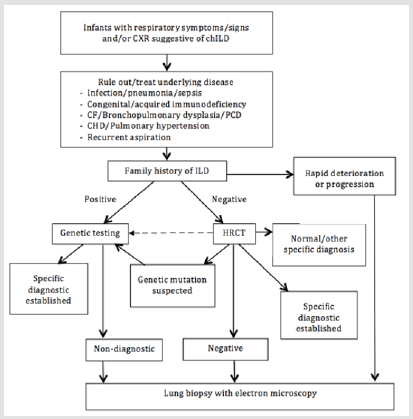
Figure 7: Proposed general diagnostic approach for childhood interstitial lung disease more prevalent in infancy. Dotted line indicate path that may be considered depending on clinical context. CF = cystic fibrosis; PCD = primary ciliary dyskinesia, CHD = congenital heart disease. This approach was adapted from American Thoracic Society [9].
NKX2.1 Mutations: NKX2.1 gene or thyroid transcription factor 1 (TTF-1) binds to specific sequences in the genes encoding SP-B, SP-C and ABCA3 to regulate their expression. Mutations of this gene result in severe respiratory distress and chronic ILD [45]. Spectrum of pulmonary presentations is varying from neonatal respiratory distress, asthma-liked symptom to pulmonary fibrosis. Not only involve in surfactant homeostasis, NKX2.1 gene also express in thyroid and central nervous system. So, patients with NKX2.1 mutation can develop hypothyroidism, developmental delay, hypotonia, seizure and choreoathetosis. These abnormalities form “brain-thyroid-lung syndrome” [46].
Treatments
Childhood interstitial lung disease is a rare condition with diverse range of etiologies. Until now, there is no specific treatment that effective for all cases and no controlled trials of therapeutic management in chILD. Treatments usually base on case reports, case series or uncontrolled studies. Main treatments can be divided to supportive and preventive care, pharmacological therapy and lung transplantation.
Supportive and Preventive Care
In every chILD infant with hypoxemia or low oxygen saturation, supplement oxygen should be provided. Growth should be evaluated and followed, if patients have growth retardation, nutritional support is indicated. Like other chronic lung diseases, harmful environmental exposures, second-hand smoker should be avoided. Furthermore, they may benefit from influenza vaccine, pneumococcal vaccine and palivizumab [47,48]. Genetic counseling should undergo for family members if the disease is associated with childhood/ adult onset ILD such as surfactant protein C mutations.
Pharmacological Therapy
Corticosteroids are the most common medical prescribed, along with hydroxychloroquine and azithromycin [49]. Corticosteroids believe to be effective due to their anti-inflammatory effect. No study declares the benefit of one steroid over another but in acute/ deteriorating conditions methylprednisolone is chosen and for long term oral prednisolone is used. The dosage of steroid may apply from other anti-inflammatory conditions. For long-term use of steroid, be aware of side effects and patients should be monitored bone density, serial growth evaluation and ophthalmologic examination [50]. However, in some chILD conditions without inflammatory process such as NEHI, steroid is not effective [51]. Hydroxychloroquine is another immunosuppressant. Response to this therapy has been reported in some cases of ABCA3 mutations and surfactant protein C deficiency [52]. Complete blood count and ophthalmologic examination also indicated if receiving long term of hydroxychloroquine [53]. The decision to start immunosuppressant depends on severity, progression of disease, comorbidities, prognosis without treatment and also family preferences [9]. Azithromycin is an antibiotic that have anti-inflammatory and immunomodulatory effects. Because of it fewer side effects, this antibiotic is used in many chILD conditions both single therapy and combined with corticosteroid or hydroxychloroquine.
Lung transplantation
Lung transplantation is the treatment of choice for end stage lung disease. Many chILD that symptoms are very severe, and prognosis is very poor such as ACDMPV, surfactant protein B deficiency or some ABCA3 mutations, lung transplantation should be considered [9]. Survival rates after transplantation in chILD are 51% and 45%, for 5 and 7-year respectively [54].
Conclusion
Interstitial lung disease composes of a large spectrum of heterogeneous disorders. ILD more common in first year of life is not similar to childhood and adult. The approach and investigation depend on onset of symptoms, underlying disease, other associated abnormalities and family history. Most of the diseases are diagnosed by lung biopsy but some can be diagnosed from CT findings and genetic sequencing. Even though mainly treatment of interstitial lung disease is supportive treatment, steroid may have role. Immunization and nutritional support are also important in affected infants. Further studies are essential to explain the pathogenesis and establish evidenced-based therapies for ILD.
Acknowledgement
None.
Disclosure statement
The authors declare that there are no potential conflicts of interest.
References
- Nogee LM (2017) Interstitial lung disease in newborns. Semin Fetal Neonatal Med 22(4): 227-233.
- Kornum JB, Christensen S, Grijota M, Pedersen L, Wogelius P, et al. (2008) The incidence of interstitial lung disease 1995-2005: A Danish nationwide population-based study. BMC Pulm Med 8: 24.
- Neil JH, Yvonne Z, Dominic F, Hiran S, Amy PM, et al. (2015) Childhood interstitial lung disease: A systematic review. Pediatr Pulmonol 50(12): 1383-1392.
- Travis WD, Costabel U, Hansell DM, King TE, Lynch DA, et al. (2013) An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 188: 733-748.
- Deutsch GH, Albright E, Chou PM, Cool CD, Coventry S, et al. (2005) Defining the spectrum of diffuse lung disease in infancy: A working classification of the pediatric interstitial lung disease cooperative. Mod Pathol 18: 304.
- Clement A (2004) ERS Task Force. Task Force on Chronic Interstitial Lung Disease in Immunocompetent Children. Eur Respir J 24: 686-697.
- Fan LL, Mullen AL, Brugman SM, Inscore SC, Parks DP, et al. (1992) Clinical spectrum of chronic interstitial lung disease in children. J Pediatr 121: 867-872.
- Fan LL, Kozinetz CA, Deterding RR, Brugman SM (1998) Evaluation of a diagnostic approach to pediatric interstitial lung disease. Pediatrics 101: 82-85.
- Kurland G, Deterding RR, Hagood JS, Young LR, Brody AS, et al. (2013) American Thoracic Society Committee on Childhood Interstitial Lung Disease (CHILD) and the CHILD Research Network. An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med. 188(3): 376-394.
- Deutsch GH, Young LR, Deterding RR, Fan LL, Dell SD, et al. (2007) Pathology Cooperative Group; CHILD Research Co-operative. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med 176: 1120-1128.
- Thacker PG, Vargas SO, Fishman MP, Casey AM, Lee EY (2016) Current Update on Interstitial Lung Diseases of Infancy New Classification System, Diagnostic Evaluation, Imaging Algorithms, Imaging Findings, and Prognosis. Radiol Clin N Am 54: 1065-
- Fan LL, Kozinetz CA (1997) Factors influencing survival in children with chronic interstitial lung disease. Am J Respir Crit Care Med 156: 939-942.
- Lum S, Stocks J, Castile R, Davis S, Henschen M, et al. (2005) American Thoracic Society; European Respiratory Society. Raised volume forced expirations in infants: guidelines for current practice. ATS/ERS statement. Am J Respir Crit Care Med 172: 1463-1471.
- Kerem E, Bentur L, England S, Reisman J, O Brodovich H, et al. (1990) Sequential pulmonary function measurements during treatment of infantile chronic interstitial pneumonitis. J Pediatr 116(1): 61-67.
- Lee EY (2013) Interstitial lung disease in infants: new classification system, imaging technique, clinical presentation and imaging findings. Pediatr Radiol. 43(1): 3-13.
- Lee EY, Cleveland RH, Langston C (2012) Imaging in pediatric pulmonology. New York: Springer: 99-154.
- Newman B, Krane EJ, Gawande R, Holmes TH, Robinson TE (2014) Chest CT in children: anesthesia and atelectasis. Pediatr Radiol 44: 164-172.
- Zucker EJ, Lee EY (2014) Radiology illustrated: Pediatr Radiol.
- Brody AS, Guillerman RP, Hay TC, Wagner BD, Young LR, et al. (2010) Neuroendocrine cell hyperplasia of infancy: diagnosis with high-resolution CT. AJR Am J Roentgenol 194(1): 238-244.
- Steve C, Adam J, Lisa RY (2019) Children’s interstitial and diffuse lung disease. Lancet Child Adolesc Health 3: 568-577.
- Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, et al. (2009) Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet 84: 780-791.
- Thouvenin G, Abou TR, Flamein F, Guillot L, Le Bourgeois M, et al. (2010) Characteristics of disorders associated with genetic mutations of surfactant protein C. Arch Dis Child 95: 449-454.
- Rock MJ (1995) The diagnostic utility of bronchoalveolar lavage in immunocompetent children with unexplained infiltrates on chest radiograph. Pediatrics 95: 373-377.
- Lee EY, Robert LC (2012) Interstitial Lung Disease in Infants and Children: New Classification System with Emphasis on Clinical, Imaging, and Pathological Correlation. Pediatr Radiol 12: 99-154.
- Lawrence MN (2017) Interstitial lung disease in newborns. Semin Fetal Neonatal Med. 22(4): 227-233.
- Langston C, Dishop MK (2009) Diffuse lung disease in infancy: A proposed classification applied to 259 diagnostic biopsies. Pediatr Dev Pathol. 12: 421-437.
- Lee EY (2013) Interstitial lung disease in infants: New classification system, imaging technique, clinical presentation and imaging findings. Pediatr Radiol 43: 3-13.
- Semple TR, Ashworth MT, Owens CM (2017) Interstitial lung disease in children made easier…well, almost. RadioGraphics 37: 1679-1703.
- Sen P, Yang Y, Navarro C, Silva I, Szafranski P, et al. (2013) Novel FOXF1 mutations in sporadic and familial cases of alveolar capillary dysplasia with misaligned pulmonary veins imply a role for its DNA binding domain. Hum Mutat. 34: 801-811.
- Szafranski P, Gambin T, Dharmadhikari AV, Akdemir KC, Jhangiani SN, et al. (2016) Pathogenetics of alveolar capillary dysplasia with misalignment of pulmonary veins. Hum Genet. 135: 569-586.
- Biko DM, Schwartz M, Anupindi SA, Altes TA (2008) Subpleural lung cysts in Down syndrome: prevalence and association with coexisting diagnoses. Pediatr Radiol 38: 280-284.
- Guillerman RP (2010) Imaging of childhood interstitial lung disease. Pediatr Allergy Immunol Pulmonol. 23(1): 43-68.
- Johnson DE, Georgieff MK (1989) Pulmonary neuroendocrine cells. Their secretory products and their potential roles in health and chronic lung disease in infancy. Am Rev Respir Dis. 140(6): 1807-
- Cutz E, Yeger H, Pan J (2007) Pulmonary neuroendocrine cell system in pediatric lung disease- recent advances. Pediatr Dev Pathol. 10(6): 419-435.
- Deterding RR (2010) Infants and young children with children’s interstitial lung disease. Pediatr Allergy Immunol Pulmonol 23: 25-31.
- Canakis AM, Cutz E, Manson D, O’Brodovich H (2002) Pulmonary interstitial glycogenosis: A new variant of neonatal inter- stitial lung disease. Am J Respir Crit Care Med. 165: 1557-1565.
- Lanfranchi M, Allbery SM, Wheelock L, Perry D (2010) Pulmonary interstitial glycogenosis. Pediatr Radiol. 40(3): 361-365.
- Castillo M, Vade A, Lim Dunham JE, Masuda E, Massarani Wafai R (2010) Pulmonary interstitial glycogenosis in the setting of lung growth abnormality: Radiographic and pathologic correlation. Pediatr Radiol 40(9): 1562-
- Paolo S, Andrew B (2016) Interstitial Lung Disease in Children Younger Than 2 Years. Pediatrics 137(6).
- Nogee LM, Garnier G, Dietz HC, Singer L, Murphy AM, et al. (1994) A mutation in the surfactant protein B gene responsible for fatal neonatal respiratory disease in multiple kindreds. J Clin Invest. 93(4): 1860-1863.
- Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF (2005) A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol. 32(6): 521-530.
- Markart P, Ruppert C, Wygrecka M, Schmidt R, Korfei M, et al. (2007) Surfactant protein C mutations in sporadic forms of idiopathic interstitial pneumonias. Eur Respir J 29(1): 134-137.
- Glasser SW, Hardie WD, Hagood JS (2010) Pathogenesis of interstitial lung disease in children and adults. Pediatr Allergy Immunol Pulmonol. 23(1): 9-14.
- Wambach JA, Casey AM, Fishman MP, Wegner DJ, Wert SE, et al. (2014) Genotype-phenotype correlations for infants and children with ABCA3 deficiency. Am J Respir Crit Care Med. 189(12): 1538-1543.
- Krude H, Schütz B, Biebermann H, Von Moers A, Schnabel D, et al. (2002) Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2-1 haploinsufficiency. J Clin Invest. 109(4): 475-480.
- Guillot L, Carré A, Szinnai G, Castanet M, Tron E, et al. (2010) NKX2-1 mutations leading to surfactant protein promoter dysregulation cause interstitial lung disease in Brain- Lung-Thyroid Syndrome. Hum Mutat 31(2): E1146-E1162.
- Harper SA, Fukuda K, Uyeki TM, Cox NJ, Bridges CB (2005) Advisory Committee on Immunization Practices (ACIP), Centers for Disease Control and Prevention (CDC). Prevention and control of influenza: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 54: 1-40.
- Fenton C, Scott LJ, Plosker GL (2004) Palivizumab: A review of its use as prophylaxis for serious respiratory syncytial virus infection. Paediatr Drugs 6: 177-197.
- Bush A, Cunningham S, De Blic J, Barbato A, Clement A, et al. (2015) European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax 70: 1078-1084.
- Ward LM (2005) Osteoporosis due to glucocorticoid use in children with chronic illness. Horm Res 64: 209-221.
- Lukkarinen H, Pelkonen A, Lohi J, Barbato A, Clement A, et al. (2013) Neuroendocrine cell hyperplasia of infancy: a prospective follow-up of nine children. Arch Dis Child 98: 141-144.
- Williamson M, Wallis C (2014) Ten-year follow up of hydroxychloroquine treatment for ABCA3 deficiency. Pediatr Pulmonol 49: 299-230.
- Chang WH, Katz BJ, Warner JEA, Vitale AT, Creel D, et al. (2008) A novel method for screening the multifocal electroretonogram in patients using hydroxychloroquine. Retina 28: 1478-1486.
- Benden C, Aurora P, Edwards LB, Kucheryavaya AY, Christie JD, et al (2011) The registry of the International Society for Heart and Lung Transplantation: fourteenth pediatric lung and heart-lung transplantation report 2011. J Heart Lung Transplant 30: 1123-1132.
