 Mini Review
Mini ReviewAbstract
Coronaviruses are RNA viruses that can infect humans and animals. The most famous representatives of the genus Betacoronavirus are SARS-CoV, MERS-CoV and the new 2019-nCoV. Today, the task of detecting a new coronavirus is urgent. We carried out a bioinformatics analysis of the available in the literature and recommended by the WHO and the governments of some country’s primer sets for real-time RT-PCR detection. Based on mutational variability of the genome of the closely related SARS-CoV virus, a map of conservative and variable regions was constructed. Two sets of primers were selected that will show themselves in the most efficient way, since they anneal not only to the conservative areas of 2019-nCoV, but also to conservative areas of SARS-CoV genome, therefore, possible mutational variation in the genome of the new coronavirus will not affect the results of the diagnosis of the disease.
Keywords: Coronavirus; Respiratory; Neurological system; Coronavirinae; Betacoronavirus
Introduction
On 31st December 2019, the World Health Organization (WHO) announced for the first-time information about several unexplained cases of pneumonia of unknown etiology in Wuhan, Hubei province, China [1]. On 7 January 2020, the genome of a new infectious agent was isolated and on 12 January 2020 WHO assigned the name of the new coronavirus as 2019 novel coronavirus (2019-nCoV). Coronaviruses are RNA viruses that damage the respiratory, hepatic, enteric and neurological systems. Coronaviruses belong to the order Nidovirales, Coronaviridae family, Coronavirinae subfamily. The Coronavirinae subfamily includes four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus and Deltacoronavirus. 2019-nCoV is a single-stranded RNA virus and belongs to the family Coronaviridae, genera Betacoronavirus. The most studied representatives of Betacoronavirus genera are SARSCoV, MERS-CoV and new 2019-nCoV. SARS-CoV was the causative agent of the acute respiratory syndrome in human population in 2002/2003. The natural reservoir of SARS-CoV are bats and intermediate hosts are camels and Himalayan civet. The SARS-CoV epidemic affected 37 countries, the number of registered cases amounted more than 8,000 and 774 were fatal (mortality rate about 10%) [2]. In 2012, another coronavirus, MERS (MERS-CoV) caused the epidemic of the Middle East respiratory syndrome. The natural reservoir of MERS-CoV are camels. 2494 cases of MERSCoV were registered since 2012 and 858 were fatal (mortality rate about 37%) [2].
The most cases are geographically associated with the Arabian Peninsula (82% of cases are reported in Saudi Arabia). Coronavirus 2019-nCoV is suspected to be a recombinant virus between bat coronavirus and a coronavirus with unknown origin. The 2019- nCoV genetic sequence is similar to the SARS-CoV sequence about 70% and to the MERS-CoV sequence about 35%. On 2 February 2020 2019-nCoV was detected in 14557 cases globally. Among them 14411 cases in China, 304 were fatal. 2019-nCoV was detected in 23 countries [3]. The purpose of this study is to compare published primer sets and primer sets recommended by WHO and ministries of health of different countries in order to select the most specific ones, which can distinguish SARS-CoV and 2019-nCoV. We conducted a comparative analysis of the genomic sequences of the 2019-nCoV relative to the SARS-CoV with further identification of the conserved and variable regions of the 2019-nCoV. We reviewed literature data to find the most effective 2019-nCoV detection method in the biological samples of the patients with the symptoms of the virus infection. Based on the data obtained, 22 sets of primers were selected for further analysis, including those recommended by WHO and ministries of health of different countries (sequences and information about primer sets are in the Table 1 of the supplement). They were chosen to detect regions of the 2019-nCoV virus genome by real-time RT-PCR: 1,2 sets of primers [4], 3,4 sets of primers [5], 5 sets of primers [2], 6,7,8,9 sets of primers [6], 10,11 sets of primers (recommended by WHO) [7], 12,13,14 sets of primers [8], 15-21 sets of primers [9], 22 set of primers [10].
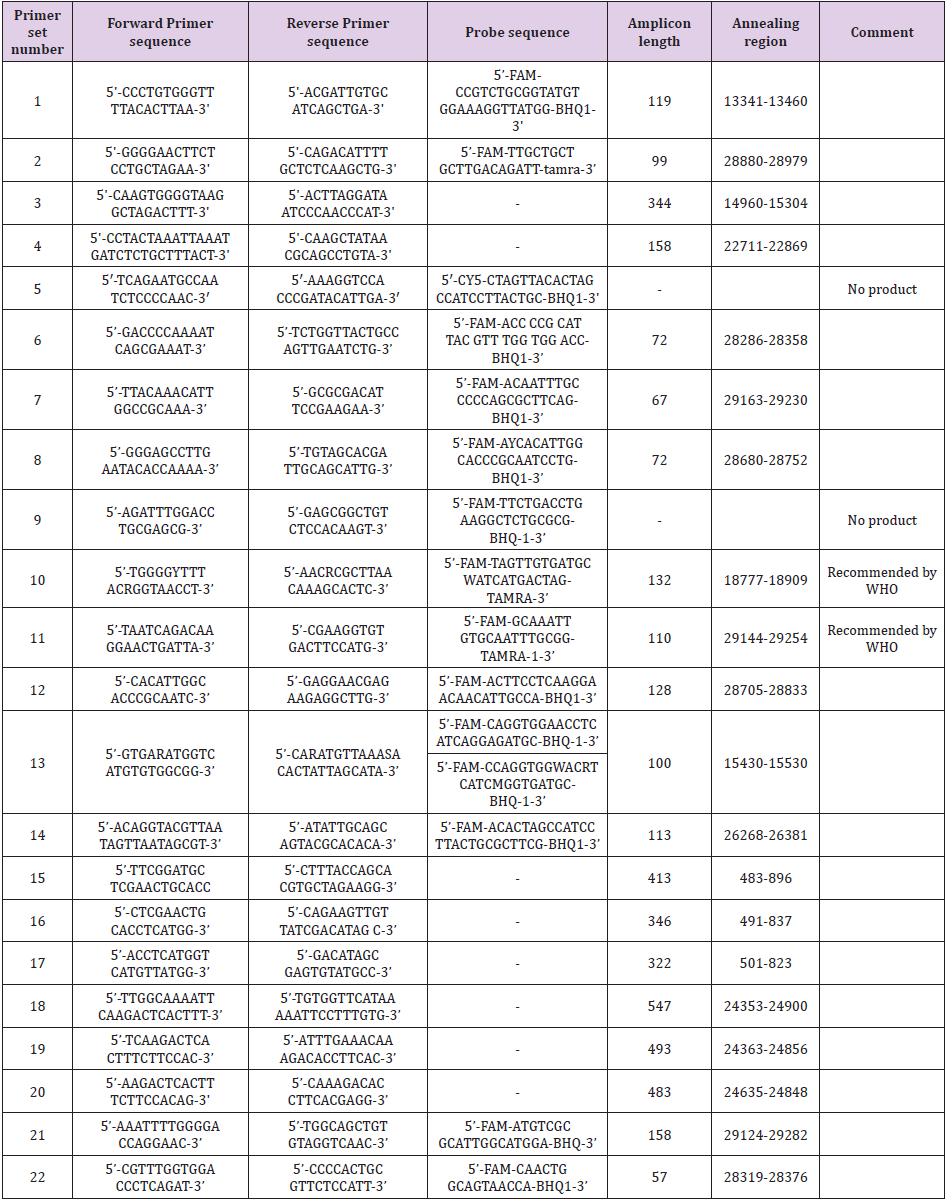
Table 1: Information about analyzed primer sets.
One of the important tasks was to identify the sets of primers that allow the most reliable detection of 2019-nCoV RNA, the genome of which can contain highly variable regions due to frequently occurring mutations. The task is proposed to be solved based on already available data on sequencing of the 2019- nCoV genome contained in the NCBI database. An analysis of the variability of the SARS-CoV genome, which is the genetically closest to the new coronavirus, can help to predict the positions of the most variable regions in the 2019-nCoV genome. At this moment, the NCBI database contains twenty 2019-nCoV nucleotide sequences, among which ten are the complete genome. Multiple alignment of these six sequences was performed by the Muscle program, and it allows to create a consensus sequence of the entire genome as a whole. For the 14 remaining sequences, the local alignment on the genome and construction of a more accurate 2019-nCoV consensus sequence were performed with Biopython library facilities. According to the results of a search of the annealing sites in the 2019-nCoVconsensus sequence for the 22 different sets of primers the most applicable were 1, 2, 6-8, 10-22 (including those recommended by WHO–10, 11) sets. Each primer from these sets has a completely complementary annealing site. The primers of the 3 and 4 sets contain no more than 2 nucleotide substitutions and amplify the product with a length of 344 and 158 nucleotide pairs, respectively. A schematic representation of the sites of annealing of primer sets is shown in Figure 1. For sets 5, 9, adequate annealing sites were not found.

Figure 1: Schematic representation of the sites of annealing primer sets.
The consensus sequence of the SARS-CoV genome was constructed on the base of the alignment by Muscle program of the 272 complete genomes contained in the NCBI database. A search for primer annealing sites for this sequence was performed. The best results were obtained for 13 and 14 sets of primers. The primers of 2, 3, 7, 8, 10-12, 22 sets contain no more than 2 nucleotide substitutions and amplify products with a length of 99, 344, 67, 72, 132, 110, 128 and 57 bp, respectively. In the case of the 1, 4-6, 9, 15-21 sets, adequate annealing sites were not found. 13 and 14 primer sets were most optimal for the detection of 2019- nCoV genome regions and the SARS-CoV consensus sequence. Therefore, these sets can be used for more reliable detection of the presented coronaviruses. Based on the data obtained, the 1, 4, 6, 15-21 sets can be used for differential detection of 2019-nCoV, because no annealing sites in the SARS-CoV genome were found for these primers.
In the case of the analysis of the consensus sequence, the most variable regions in the SARS-CoV genome were revealed. One region from 21489 to 23837 nucleotide pairs corresponds to the first half of the S gene, the other region from 27922 to 28294 nucleotides corresponds to the ORF8 gene. The new 2019-nCoV coronavirus contains the S gene, which, probably, like the similar gene in SARSCoV, can be highly variable from the 5’end. Therefore, the design of primers to other potentially more conservative parts of the genome may be more reliable.
Conclusion
Based on a comparative analysis of the 2019-nCoV and SARSCoV genomic sequences, potential variable regions of the 2019- nCoV virus genome were identified. Based on the data obtained, it can be assumed that despite the potential mutational variation in the 2019-nCoV virus genome, the most optimal are 13 and 14 primer sets, since they anneal to conserved regions of the genome of the closely related SARS-CoV virus. Based on the analysis, we identified primer sets that can be used for screening diagnostics, and to confirm the diagnosis.
References
- (2020) Coronavirus. World Health Organization WHO.
- Chaolin Huang, Yeming Wang, Xingwang Li, Lili Ren, Jianping Zhao, et al. (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The lancet 395(10223): 497-506.
- (2020) Novel Coronavirus (2019-nCoV) Situation Report-13. WHO?
- Na Zhu, Dingyu Zhang, Wenling Wang, Xinwang Li, Bo Yang, et al. (2020) A Novel Coronavirus from Patients with Pneumonia in China, 2019. 382: 727-733.
- Jasper Fuk Woo Chan, Shuofeng Yuan, Kin-Hang Kok, Kelvin Kai-Wang To, Hin Chu, et al. (2020) A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 395(10233): 514-523.
- (2020) Real-time RT-PCR panel for detection 2019-novel coronavirus. Instruction for Use Department of health & human services, Centers for Desease control and prevention (CDC).
- Leo Poon, Daniel Chu, Malik Peiris (2020) Detection of 2019 novel coronavirus (2019-nCoV) in suspected human cases by RT-PCR. HKU MED.
- Corman Victor M, Landt Olfert, Kaiser Marco, Molenkamp Richard, Meijer Adam, et al. (2020) Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill 25(3).
- Naganori Nao, Kazuya Shirato, Harutaka Katano, Shutoku Matsuyama, Makoto Takeda (2020) Detection of second case of 2019-nCoV infection in Japan. National Institute of Infectious Diseases 1-23.
- (2020) RT-PCR protocol for the detection of 2019-nCoV. Diagnostic detection of Novel coronavirus 2019 by Real time RT-PCR Department of Medical Sciences, Ministry of Public Health.
