 Research Article
Research ArticleAbstract
Background: Normokalemic periodic paralysis (Normo KPP) of skeletal muscle is an autosomal dominant disorder caused by mutations in the calcium voltage-gated channel subunit (CACNA1S) or sodium voltage-gated channel subunit 4 (SCN4A) genes, which lead to ion-channel dysfunction. Little is known about the relationship between mutation genotypes and the clinical symptoms of Normo KPP.
Methods: To examine the mutation status of CACNA1S and SCN4A, in a Chinese family segregating NormoKPP with atypical clinical symptoms, we performed a series of clinical examinations and genetic analyses with whole-exome sequencing.
Result: Whole-exome sequencing revealed a substitution of c.2111C>T in SCN4A, resulting in the amino acid substitution p.T704M in all affected family members.
Conclusion: The present study supports a causative role for this mutation in SCN4A in NormoKPP and provides information about the relationship between genotype and atypical clinical symptoms.
Keywords: Normokalemic; Periodic Paralysis; Ion Channel; Mutation; SCN4A; CACNA1S
Introduction
Periodic paralysis (PP) is a group of ion-channel dysfunction diseases [1]. Based on serum potassium level, PP can be classified into hypokalemic periodic paralysis (Hypo KPP), hyperkalemic periodic paralysis (Hyper KPP), and normokalemic periodic paralysis (Normo KPP). The term Normo KPP was originally used in the 1960s [2] to describe a very rare disease characterized by repeated attacks of flaccid muscle weakness or paralysis, despite normal serum potassium levels. Trigger factors for these episodes include rest after acute exercise, cold, hunger, emotional tension, carbohydrate-rich meals, and potassium supplements. Concerns episodes of paralysis is most in the early morning or during breaks from vigorous activities [3,4]. Several familial and sporadic cases of this disease have been reported [5,6]. Pathogenic mutations in exons 12, 13, 18, and 24 of SCN4A, leading to the amino acid changes T704M, R675G, R675W, R675H, R675Q, R1129Q, and M1592V, have been reported to cause Normo KPP in patients from China and other countries [5-8]. Compared with other diseases causing PP, Normo KPP has no specific clinical features, with electromyography, electrocardiogram, blood sugar, cerebrospinal fluid, and routine biochemical laboratory and auxiliary examinations generating normal results during an attack.
In this study, we adopted the Ion Torrent Proton sequencing platform for whole-exome scanning, which has advantages over Sanger sequencing in terms of read throughput, cost, operation time, and potential for use as a standardized procedure in hospital. Given the diverse clinical symptoms of Normo KPP, the scope of the investigation was expanded to the whole-exome scale, to obtain comprehensive information on the genetics underlying the pathogenic features of the condition. We characterized a Chinese family with atypical Normo KPP features and identified a classical c.2111C>T (p.T704M) mutation in SCN4A that is predicted to be responsible for the disease.
Patients and Methods
Patients and Families
A Chinese family of 29 members with Normo KPP was recruited from the city of XX, China (Figure 1). The proband was a 32-yearold man with a history of recurrent muscle weakness and gait disturbance, who was hospitalized in 2015. The age of initial onset was 8 years. The common clinical features affecting living 16 family affected members were as follows (Table 1): episodic paralysis of the limbs with concomitant normal serum potassium concentration; onset at 7–10 years old; patients could stand but not walk, and could lift their upper limbs, but found it difficult to hold them; no dyspnea or dysphagia and no episode of painful cramps and stiffness in limbs; examination of electrocardiogram , thyroid function, and muscle enzymes generated normal results; performance of electromyogram showed no myotonic discharge or increased insertion activity. The patients awake paralyzed the morning following vigorous exercise and cold temperatures. Significantly atypical feature from other Normo KPP families [6- 9] was that treatment with large doses of physiological saline had no effect, whereas calcium gluconate was effective. In addition, acetazolamide could reduce the number of attacks and alleviate attack symptoms.
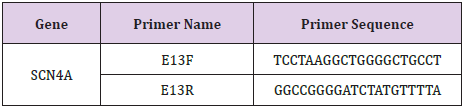
Table 1: PCR primers used for SCN4A exon 13 screening.
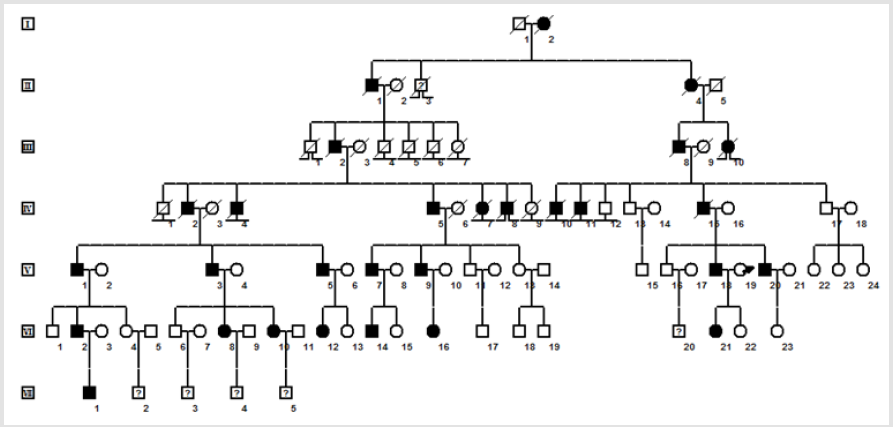
figure 1: Family pedigree. Note: Black symbols, affected individuals; open symbols, unaffected individuals; male, square; female, circle; question marks, unknown phenotype; ⊥ non-offspring; lost non-offspring; lost to follow-up; slash mark, deceased individuals; arrow, proband.
Detailed records of the patients’ medical histories, including physical examinations, blood potassium levels and results of electromyogram were obtained during attacks. The diagnosis of Normo KPP was made based on symptoms, physical signs, and normal blood potassium levels range during attacks. All participants in this study were enrolled and evaluated at the First Affiliated Hospital of Bengbu Medical College. Informed consent was obtained from participants under the research protocols approved by the Bengbu Medical College ethics review board (BYYFY-2012KY04).
DNA Extraction, Ion Torrent Proton Library Preparation, and Sequencing
Peripheral anticoagulated whole blood samples were collected from four family members, including a father (V-18; affected), mother (V-19; unaffected), and two daughters (one affected and one unaffected; VI-21 and VI-22, respectively), and genomic DNA was extracted using a commercial Blood Genomic DNA Miniprep kit (Axygen, Union City, CA, USA). The Qubit dsDNA HS (High Sensitivi ty) Assay kit was used to quantify DNA with the Qubit® Fluorometer (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA), according to the manufacturer’s instructions. Ion Torrent Proton adapter-ligated libraries were generated for each of the four family members using the Ion AmpliSeq Library Kit 2.0 (Termofisher, Cat.no.4476610), following the manufacturer’s protocol. Following AMPureXP bead purification (Beckman Coulter), the concentrations of the libraries were determined by qPCR using an Ion Library Taqman Quantitation Kit (Termofisher, Cat. No. 4468802). Emulsion PCR and sample enrichment were performed using the Ion PI HI-Q Template OT2 200 Kit v2 (Termofisher, Cat.no.A26434), according to the manufacturer’s instructions. Isolation of templated on the Ion Sphere Particles (ISPs) was performed using Ion OneTouch ES (Termofisher, Guilford, CT, United States).Template-positive of ISPs were enriched and sequencing was performed using an Ion PI HI-Q Sequencing 200 Kit v2 (Termofisher,Cat.no.A26433) on the Ion Torrent Proton instrument (Termofisher, Guilford, CT, United States).
Bioinformatics Analyses
Raw data from Ion Torrent Proton runs were processed using the Ion Torrent platform-specific pipeline software, Torrent Suite v4.6 (Life Technologies) to generate sequence reads, for sequence alignment, and to extract single nucleotide variants and indels. Initial variant calling was performed using the Ion Torrent platform-specific pipeline software with the plug-in ‘‘variant caller” program. Two annotation steps were used to obtain information about gene mutations associated with disease. Torrent Suite Variant Caller (TSVC; version 4.6) plug-in generated files were filtered and annotated using ANNOVAR software [10], then the variant caller format (VCF) files generated by ANNOVAR were further filtered and annotated using WANNOVAR software [11]. LOVD database and PubMed literature queries were performed to determine whether variants had been reported as pathogenic and identify disease loci; suspected pathogenic mutations were verified by Sanger sequencing. SCN4A sequences of five non-human species for homology comparisons were obtained using NCBI BLAST and alignments were carried out using Clustal W to check conservation.
Sanger Sequencing
Mutation in candidate gene SCN4A exon 13 (SCN4A, NM_000334.4) were confirmed by Sanger sequencing. Primers were designed using Primer Premier 5.0 (SCN4A E13F- TCCTAAGGCTGGGGCTGCCT, SCN4A E13RGGCCGGGGATCTATGTTTTA). Amplified fragments were sequenced on an ABI Prism 3730XL DNA analyzer (Applied Biosystems) using a Big Dye Terminator Cycle Sequencing Kit v2 (Applied Biosystems). Chromatograms illustrating the Sanger sequencing results were analyzed using the SeqMan Pro application in DNASTAR’s Lasergene Molecular Biology Suite software.
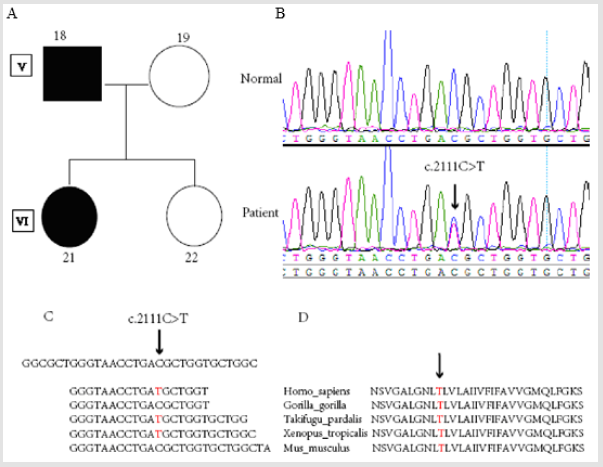
figure 2: SCN4A mutation detection in a family with normokalemic periodic paralysis. Note: A. Pedigree structure of a NormoKPP family carrying the c.2111C>T mutation in SCN4A. B. SCN4A DNA sequencing results of the patient and a healthy control. C. SCN4A Ion torrent DNA sequencing results for the patient. D. Sequence conservation of amino acid 704 in the SCN4A protein.
We performed whole-exome sequencing of a four-member Chinese family including one parent and one child affected with Normo KPP, resulting in > 100× coverage (Figure 2A). On average, 75 million high quality reads (AQ ≥ 20) were generated per run. The average depth of total coverage was 148x, and the minimal coverage was 28x. Candidate variants (n = 94) (supplementary data) were selected after mapping to the reference genome and filtering using relevant databases. Finally, one variant (SCN4A, p.T704M) was identified in a gene of interest (Figure 2C). The result of exome sequencing was verified by Sanger sequencing (Figure 2B) in affected patients (IV5, V1, V3, V5, V7, V9, V18, V20, VI2, VI8, VI10, VI12, VI14, VI16, VI21, VII1). The threonine residue at position 704 of SCN4A is highly conserved among humans, mice, gorilla, Fugu, and Xenopus (Figure 2D).
Discussion
SCN4A encodes the a-subunit, voltage-gated sodium channel, Nav1.4 whose structure consists of four domains (DI-DIV) of six transmembrane helical segments (S1–S6). Here, we report a pathological mutation in the SCN4A gene in a family presenting with hereditary Normo KPP. In the present study, whole-exome and Sanger sequencing identified C/T heterozygosity in the SCN4A gene at nucleotide position 2111 in the 13th exon. Both of the affected members in the family carried the p.T704M mutation, whereas the unaffected members did not. Pathophysiological studies show the mutation conduct gating pore current in both activated and slowinactivated states, which would cause increased influx of sodium near the resting membrane potential, membrane depolarization, sodium overload, action potential failure [12]. Structural studies elucidate the molecular mechanism by which mutations in S4 cause pathogenic gating pore currents and suggest that ion permeation through the gating pore is controlled dynamically by the state of the voltage sensor and by rotamer conformations of R4 [13].Patients of the family exhibited Normo KPP with atypical features, in that treatment with high dose physiological saline was ineffective, while calcium gluconate was effective. Different families with the p.T704M mutation of SCN4A showed various clinical picture. The mutation p. T704M identified in family accounts for the majority of patients with hyper KPP [14,15]. Some patients harbouring this mutation also exhibit periodica paramyotonia and paramyotonia congenita [16,17]. Some of families diagnosed as Normo PP were subsequently found to have the Hyper PP mutations T704M or M1592V, leading to the suggestion that Normo PP may be a phenotypic variant of HyperPP [18]. Patients with Normo PP may experience transient high serum potassium level before attacks. Why Genotype-phenotype correlations for SCN4A mutations are not straightforward? A potassium channel gene such as KCNE3, in which mutations are a rare cause of hypokalaemic or hyperkalaemic periodic paralysis, is an example of a possible modifying gene [19]. It now appears that Ca2+activated-K+-(BK) channel encoded by one gene (slo1/KCNMA1) may have relevance in disorders associated with abnormal K+ ion homeostasis including periodic paralysis and myotonia [20,21].
Saline solution is first choice for acute management of Normo KPP. The dose of saline solution usually needs more than three thousand milliliter a day and patiens with NormoKPP regularly recovered within 2-3 days. Patients of the family with Normo KPP showed no response to saline solution, intravenous calcium gluconate was chosen to treat patients according to protocol of Neurology textbook [22].We presumed the mechanism of which was calcium gluconate enhance opening of calcium-activated K channels[20,21]. The prophylactic therapy with diuretics is effective in treating Normo KPP. Acetazolamide was empiric treatment for NormoKPP, the mechanism of action is unclear. Another diuretics hydrochlorothiazide was confirmed being effective in the prophylactic treatment of normoKPP caused by the SCN4A mutation of p. Thr704Met [23]. The results of this study demonstrate potential new clinical features of PP associated with p.T704M mutation in SCN4A. In addition, we also demonstrate the feasibility of next generation sequencing for mutation detection in a complex monogenic disease.
Acknowledgment
This work was supported by Anhui University of Science and Technology (KJ2013A189).
Conflict of Interest
We have no disclosure to make that qualifies as a conflict of interest.
References
- Lehmann-Horn F, Küther G, Ricker K, Grafe P, Ballanyi K, et al. (1987) Adynamia episodica hereditaria with myotonia: a noninactivating sodium current and the effect of extracellular pH. Muscle Nerve 10(4): 363-374.
- Jurkat-Rott K, Holzherr B, Fauler M, Lehmann-Horn F (2010) Sodium channelopathies of skeletal muscle result from gain or loss of function. Pflügers Archiv 460(2): 239-248.
- Bulman DE, Scoggan KA, van Oene MD, Nicolle MW, Hahn AF, et al. (1999) A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology 53(9): 1932- 1936.
- Fontaine B, Vale-Santos J, Jurkat-Rott K, Reboul J, Plassart E, Rime CS, et al. (1994) Mapping of the hypokalaemic periodic paralysis (HypoPP) locus to chromosome 1q31-32 in three European families. Nat Genet6(3): 267-272.
- Vicart S, Sternberg D, Fournier E, Ochsner F, Laforet P, et al. (2004) New mutations of SCN4A cause a potassium-sensitive normokalemic periodic paralysis. Neurology 63: 2120-2127.
- Xiuhai G, Weiping W, Ke Z, Hongbin W, Yiling S, et al. (2008) Mutations of sodium channel alpha-subunit genes in Chinese patients with normokalemic periodic paralysis. Cell Mol Neurobiol 28(5): 653-661.
- Rüdel R, Ricker K, Lehmann-Horn F (1993) Genotype-phenotype correlations in human skeletal muscle sodium channel diseases. Arch Neurol 50(11): 1241-1248.
- Hong D, Luan X, Chen B, Zheng R, Zhang W, et al. (2010) Both hypokalemic and normokalemic periodic paralysis in different members of a single family with novel R1129Q mutation in SCN4A gene. J NEUROL NEUROSUR PS 6(6): 703-704.
- Xiuhai G, Weiping W, Yanhua Z, Jianping J, Ke Z (2004) V781I in SCN4A gene exists in Chinese patients with normokalemic periodic paralysis. Chinese Journal of Medical Genetics 21: 566-569.
- Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16): e164.
- Chang X, Wang K (2012) wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet 49(7): 433-436.
- Sokolov S, Scheuer T, Catterall WA (2008) Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc Natl Acad Sci U SA 105(50): 19980 -19985.
- Jiang D, Gamal El-Din TM, Ing C, Lu P, Pomès R, et al. (2018) Structural basis for gating pore current in periodic paralysis.Nature 557 (7706): 590-594.
- Miller TM, Dias da Silva MR, Miller HA, Kwiecinski H, Mendell JR, et al. (2004) Correlating phenotype and genotype in the periodic paralyses. Neurology 63(9): 1647-1655.
- Lee YH, Lee HS, Lee HE, Hahn S, Nam TS, et al. (2015) Whole-Body Muscle MRI in Patients with Hyperkalemic Periodic Paralysis Carrying the SCN4A Mutation T704M: Evidence for Chronic Progressive Myopathy with Selective Muscle Involvement. J Clin Neurol 11(4): 331-338.
- Kim J, Hahn Y, Sohn EH, Lee YJ, Yun JH, et al. (2001) Phenotypic variation of a Thr704Met mutation in skeletal sodium channel gene in a family with paralysis periodica paramyotonica. J Neurol Neurosurg Psychiatry 70(5): 618- 623.
- Brancati F, Valente EM, Davies NP, Sarkozy A, Sweeney MG, et al. (2003) Severe infantile hyperkalaemic periodic paralysis and paramyotonia congenita: broadening the clinical spectrum associated with the T704M mutation in SCN4A. J Neurol Neurosurg Psychiatry 74(9): 1339-1341.
- Chinnery PF, Walls TJ, Hanna MG, Bates D, Fawcett PR (2002) Normokalemic periodic paralysis revisited: Does it exist? Ann Neurol 52(2): 251-252.
- Abbott GW, Butler MH, Bendahhou S, Dalakas MC, Ptacek LJ, et al. (2001) MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell 104(2): 217- 231.
- Tricarico D, Mele A, Calzolaro S, Cannone G, Camerino GM, et al. (2013) Emerging Role of Calcium-Activated Potassium Channel in the Regulation of Cell Viability Following Potassium Ions Challenge in HEK293 Cells and Pharmacological Modulation.PLoS One 8(7): e69551.
- Dinardo MM, Camerino G, Mele A, Latorre R, Conte Camerino D, et al. (2012) Splicing of the rSlo Gene Affects the Molecular Composition and Drug Response of Ca2+-Activated K+ Channels in Skeletal Muscle.PLoS One 7(7): e40235.
- Weizhi W (2001) Neurology, People's Medical Publishing House.
- Akaba Y, Takahashi S, Sasaki Y, Kajino H (2018) Successful treatment of normokalemic periodic paralysis with hydrochlorothiazide. Brain Dev 40(9): 833-836.
