 Research Article
Research ArticleAbstract
We propose a model of global permanent ischemia on rats, obtained through decapitation of the animals, in which successive metabolic examinations have been conducted at different ischemia times up until a maximum of 2 hours. In order to verify the protective role of temperature’s reduction, metabolic examinations have been conducted at three different temperatures: normothermia (37ºC); moderate hypothermia (34ºC); and low hypothermia (31ºC). For each different temperature examined the values are the results of the average of 6 animals, this to limit the alterations deriving from the unusual behavior of single specimen, consequence of the inter-subject variability inside the same species. The reference values were obtained from the brain metabolic examination of 6 rats carried out immediately after the decapitation process. The metabolic inquiries carried out were focused on the analysis of the changes of the cellular energetic metabolism through the dosage of main energy molecules (ATP and GTP), of their intermediate catabolites (ADP, AMP, GDP, GMP) and of their final products (hypoxanthine, xanthine, uric acid, adenosine and inosine); on the mitochondrial alterations, through the note of the modifications of the nicotinic coenzymes entailed in the oxidative phosphorylation process (NAD and NADP) and on the action of the main mediators of the ischemic damage. From the analysis of the results presented above, in our experimental model the result is that in conditions of global ischemia and normothermia there is a progressive reduction of the main energetic metabolites
Keywords: Ischemia; Hypothermia; Metabolism; Neuroprotection
Introduction
Despite the huge amount of literature on cerebral metabolism
and cerebral ischemia, a clear definition of ischemic cell death
universally accepted is yet to be found. Furthermore, there is
no unanimous agreement on the timing of neuronal cells death
secondary to an ischemic insult. Several studies have shown
that distinct neuronal populations have a different vulnerability
to ischemia [1-3]. There are reports in the literature on how a
5-minute global ischemia in the gerbil cause delayed death in
almost all pyramidal cells of CA1 (Ammon’s horn), without any
effect detrimental in other neuronal populations, while 20 minutes
of ischemia are sufficient to induce cell death in CA3 and do not
create damage in the hippocampal dentate granule cells [4,5].
Other studies have also shown that the same threshold of tolerance
of a neuronal population might be changed allowing recovery of the
ability of protein synthesis or interrupting the dissolution process
of intracellular organelles [6,7].
Alongside with the progresses in analyzing the pathogenesis
behind the ischemic insult, neuroprotection mechanisms have
become matter of interest. In particular studies on all those
methods, pharmacological, physical or chemical that can reduce
or delay the deleterious effects of the absence of blood flow were
considered. Among those neuroprotective measures, hypothermia
plays an extremely important role. Numerous studies have shown
that even small reductions in brain temperature induced during or after the ischemic insult, provide adequate neuroprotection [8-
13]. Hypothermia has now assumed a prominent role in studies
on ischemic brain and it is for this reason that a large part of this
work will be devoted to the analysis of the effects of the reduction
in brain temperature, even in light of our experience.
The model that we propose in this experimental study is a
situation of global permanent ischemia on rats, obtained through
the decapitation of the animals on which the successive metabolic
examinations have been conducted at different ischemia times up
until a maximum of 2 hours. In order to verify the protective role
of the temperature’s reduction along the ischemic process, the
metabolic examinations have been conducted at three different
temperatures: normothermia (37ºC); moderate hypothermia
(34ºC); and low hypothermia (31ºC). In order to reduce the
variability of the analysis deriving from the unusual behavior
of single specimen, consequence of the inter-subject variability
inside the same species 6 animals were analyzed. The reference
values were obtained from the brain metabolic examination of 6
rats carried out immediately after the decapitation process (time
0’). Metabolic analysis was focused on the sampling of the changes
of the cellular energetic metabolism through the dosage of main
energy molecules (ATP and GTP), of their intermediate catabolites
(ADP, AMP, GDP, GMP) and of their final products (hypoxanthine,
xanthine, uric acid, adenosine and inosine). Furthermore,
analysis on the mitochondrial alterations, through the note of the
modifications of the nicotinic coenzymes entailed in the oxidative
phosphorylation process (NAD and NADP) and on the action of
the main mediators of the ischemic damage, represented by the
free radicals whose activity was indirectly measured through the
modifications of the malondialdehyde levels (MDA), resulting from
the action of those substances with the membrane’s phospholipids,
just like those of the main cellular antioxidant (ascorbic acid).
Methods and Materials
Preparation of the Isolated Rat Brain
Male adult rats were anesthetized through the intraperitoneal administration of Ketamine (300mg/kg of body weight) and, after the injection of 1000 UI of heparin in the caudal vena cava, were decapitated in a way to allow the reproduction of a total ischemic state, without the influence of collateral circles. The whole head of the animals was then put in special containers for the subsequent thermic treatment. Three groups of six specimen each were put in incubator, in controlled humidity conditions, at the constant temperature of 37ºC for the following times: 30 minutes, 60 minutes, and 120 minutes. The same temporal kinetic was followed also for two more series of three groups of six specimen each put at the constant temperatures of 34 and 31ºC respectively.
In order to provide a control group of brains, six anesthetized and decapitated animals were used and immediately processed for the biochemical examinations. At the end of each period of thermic treatment, the brains were immediately immersed in liquid nitrogen and processed to obtain a deproteinized tissular extract, to be used for the definition of the biochemical parameters selected through high-performance liquid chromatography (HPLC) technique. The brains were taken from the liquid nitrogen and immediately homogenized for 60 seconds in the presence of perchloric acid (HCIO4) 1.2 M cold, so to obtain the complete deproteinization of the specimen. The Homogenate of each specimen obtained in the abovementioned way has been centrifuged for 15 minutes at 13000rpm (at 4°C); the supernatant was separated and stored at 4°C, while the pellet obtained was newly homogenized with HCIO4 1.2M cold. After a further centrifugation (13000rom/10 minutes, 4°C), the new supernatant was added to the previous of each specimen and the whole was neutralized through the addition of K2CO3 5M. Following a new centrifugation (13000 rpm/10 minutes, 4°C), the pellet of sunken salt was discarded and an equal volume of chloroform (1:1; v:v) was added to the neutralized tissular extract, in order to obtain the removal of all the non-water-soluble components present in the specimen. After having whirled the specimen for 60 seconds, a centrifugation of the specimen at 10000 rpm for 10 minutes at 4°C was carried out; the superior aqueous phase thus obtained was drawn (composed then exclusively by the aqueous supernatant free from any lipoid leftover) which was stored at -80°C until the time of the HPLC examination, before which the specimen was accurately filtered with a 0.45μM MILLIPORE-HV filter.
Biochemical Parameters of the Research
On the neutralized acid tissular extracts of rat brain, the following concentrations were evaluated through HPLC: the high energy of the phosphorylated compounds (ATP, ADP, AMP, GTP, GDP, GMP, UTP, UDP, UMP), of the nicotinic coenzymes (NAD and NADP), of the ox purine (hypoxanthine, xanthine, uric acid), of the nucleosides (adenosine and inosine), the antioxidant ascorbic acid and of the malondialdehyde (MDA) as biochemical index of lipoid peroxidation induced by the oxygen radicals (ROS) on the polyunsaturated fat acids of the phospholipids of the biological membranes.
HPLC Chromatographic Method of Examination
HPLC consists in a small modification of the previous ionic coupling methods perfected in our labs, during which the tetrabutylammonium is used as a coupling agent. A rate of the aqueous phase (100μl) will be examined through high performance liquid chromatography (HPLC) whose apparatus consist of a double pump system ThermoQuest Constametric 3500 (ThermoFinnigan Italia, Rodano, Milano, Italia) connected to an in series-diode spectrophotometric developer Spectra System UV6000LP (ThermoFinnigan Italia) which was set between the wave lengths of 190 and 330 nm. All this system is, in turn, connected to a PC for the acquisition and analysis of data, which uses a software system supplied by the same company that produces the HPLC system. For the analysis of the specimen an analytic chromatographic column Kromasil C-18, 250 mm x 4.6 mm was used, with 5μM pores (Eka Chemicals, AB, Bohus, Sweden) provided with the specific precolumn. The column was balanced with a mobile phase (A tampon) containing Hydroxide tetrabutylammonium 10 nM, KH2PO4 10nM, and methanol at 0.25%, with 7.00 pH. A gradient “with leaps” was carried out through a second tampon (B) containing hydroxide tetrabutylammonium 2.8 nM, KH2PO4 100mM, and methanol at 30%, with 5.50 pH; the gradient used is the following: 15 minutes at 100% of A; 5 minutes at 90% of A; 5 minutes at 70% of A; 15 minutes at 63% of A; 15 minutes at 55%; 3 minutes at 50% of A; 32 minutes at 48% of A; 10 minutes at 28% of A; 15 minutes at 0% of A. The flux of the chromatographic runs was 1.2 ml/min and the temperature were maintained constant at 23°C, through a specific thermostatic system. The separation of the compounds in the experiment, which are identified through the confrontation of runs of ultra-pure standards both of the respective retention times and of the absorbing spectrums, has permitted the calculus of the concentrations carried out at the specific wave lengths of each substance.
Statistical Analysis of Data
The results of the different experiments were analyzed with specific statistical tests as the two-tailed Student test-t for noncoupled specimen.
Results
The results emerged from the experiments refer to the rat groups that underwent the model of global ischemia at the temperature of 37, 34 and 31°C. The values represent the average of the groups of 6 rats at different observation times (30’-60’-120’). The values reported for time 0’ are also the average of 6 animals that underwent at the metabolic investigations immediately after the decapitation process; they then represent the benchmark controls for each group. Room temperature is the value to be considered in this case. Detailed statistical analysis has been carried out only for the parameters correlated to the main cellular cycles. At 37°C, as we move away from the beginning of the ischemia, a progressive reduction of the energetic metabolites (Figure 1; Tables 1- 5) can be observed. The data is confirmed by the parallel increase of those that can be considered the intermediate catabolites of the main cellular energetic molecules (ADP, AMP, GDP, GMP) and of their final catabolic products (hypoxanthine, xanthine, and adenosine). Also, the uric acid presents a net increment at 60’ and 120’ in comparison to the concentrations of the start: this substance is the final product of the action of the xanthine oxidase on the ox purines, described above in relation to the ways of formation of the free radicals. In turn, the gradual increase of MDA (Figure 2), index of the action of the free radicals on the membrane phospholipids, seems to perfectly correlate with the progressive reduction of the Ascorbic Acid which represents the main cellular antioxidant (Figure 3). Finally, the reduction of the concentrations of the nicotinic coenzymes (NAD and NADP) is index of the progressive alteration of the mitochondrial function, since the lower availability of these electron acceptors entails the gradual block of the mitochondrial respiratory chain and thus of the cellular oxidative phosphorylation. It is important to notice that, following the extraction of the specimen in percloric acid, there is the total transformation of the compounds reduced in the correspondent oxidative form. Hence, in the particular case of the nicotinic coenzymes it is indicated with the acronym NAD (or NADP) the total concentration of NAD+NADH (or NADP + NADPH). It is interesting to note that the reduction of NAD levels (Figure 4) could be correlated to the parallel increase of the concentrations of ADP ribose (Figure 5) which, in turn, is the expression of the activity of the poly (ADP-ribose) polymerase (PARP) system. The correlations of the two occurrences will be examined in the discussion phase.
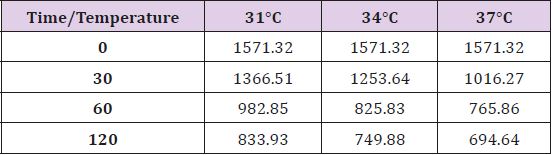
Table 1: ATP levels at different observation times and temperature.
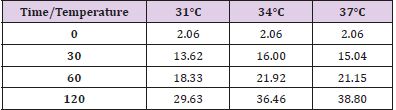
Table 2: MDA levels at different observation times and temperature.
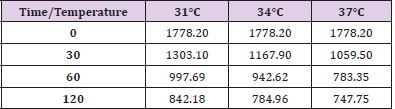
Table 3: Ascorbic acid levels at different observation times and temperature.
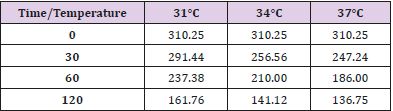
Table 4: NAD levels at different observation times and temperature.
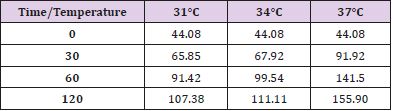
Table 5: ADP levels at different observation times and temperature.
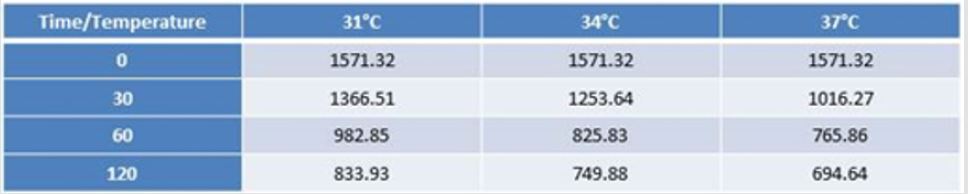
Figure 1: ATP levels at different observation times and temperature.
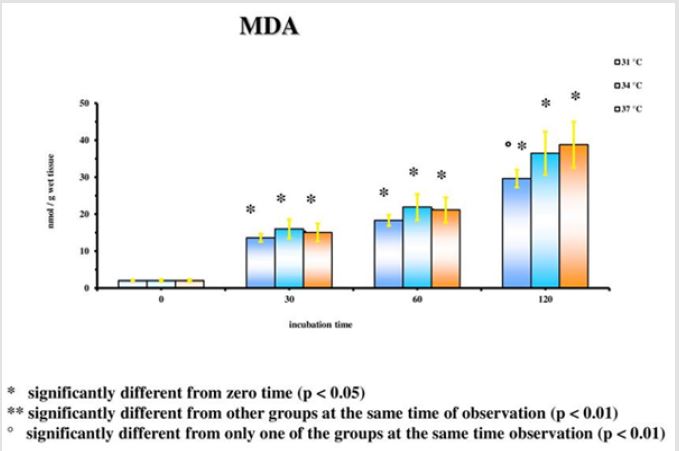
Figure 2.
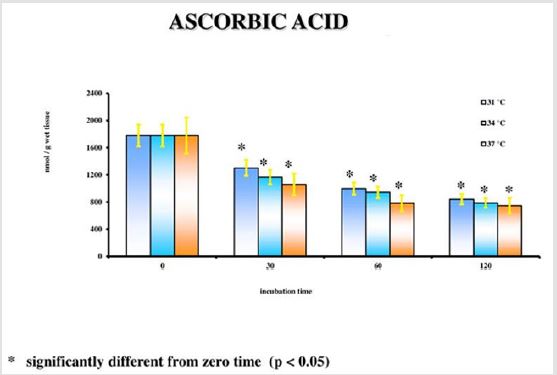
Figure 3.
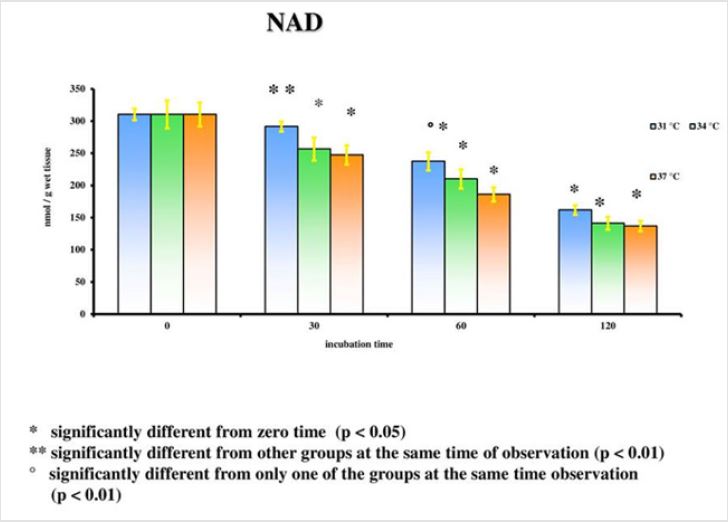
Figure 4.
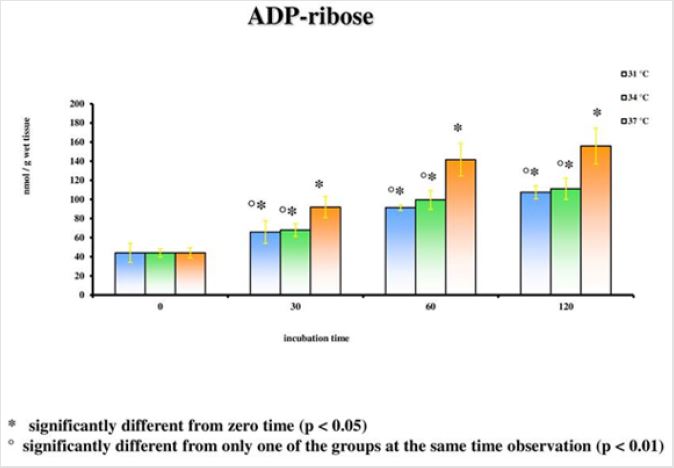
Figure 5.
In conditions of low (34°C) and moderate (31°C) hypothermia, we note the occurrence of a beginning (30’) slowdown of the ATP depletion (Figure 1), the only statistically significant value compared to the control (time 0’) is indeed the one of the group of animals at 37°C (p<0.01). After 60 minutes, all the groups present statistically different values compared to the benchmarks, index of a significant reduction of the ATP concentrations; nevertheless, the group at 31°C shows a higher conservation of the energetic metabolite which results statistically meaningful compared to the other groups for the same observation time (p<0.05). Finally, after 120 minutes of ischemia there is no observation of significant differences between the three examined groups and obviously this confirms the self-evident reduction of the ATP concentration compared to the benchmarking values. The increase of the final catabolic products ATP and GTP, represented by the sum of the oxypurines (hypoxanthine, xanthine, and uric acid) and by the sum of the nucleosides (adenosine and inosines) mirrors the same trend, resulting only modestly inferior for the groups of hypothermic rats compared to those at 37°C. In the case of the MDA (Figure 2), the concentrations of the substance progressively tend to increase in a significant manner compared to the benchmarking values, in all the groups and for all the observation times (p<0.01). Important modifications do not appear to be observed between the group of normothermic animals, the ones at 34 and 31°C for the observation times at 30 and 60 minutes. After 2 hours of ischemia, nevertheless, the group of rats at 31°C displays a minor increase of the MDA levels, which results statistically significant in relation to the group at 37°C (p<0.05) for the same observation time, but not compared to the group of animals at 34°C. The ascorbic acid behaves in a similar way, it appears only moderately more conserved at 31°C, compared to what it is observed in the normothermic animals, for all the observations times (Figure 3). The mitochondrial function appears partially maintained in condition of moderate hypothermia (31°C), as shown, in the different observation times, by the lower reduction of the NAD levels (Figure 4). Nevertheless, the mitochondrial alteration remains significant for all the groups at the different times of ischemia (p<0.01).
Notwithstanding this, the group of rats at 31°C presents a preservation of NAD levels which results statistically significant compared to the group at 37°C for all the observation times and compared to the group at 34°C for the observations at 30 and 120 minutes (p<0.05). To the contrary, significant differences between the group at 34°C and the one at 37°C are never observed. In a parallel way also the ribosylation process of the ADP results remarkably more controlled (Figure 5). Statistically significant differences for the different observation times (p<0.05) are observed between the groups at 31°C and at 34°C on one hand, and the one at 37°C on the other; the increase of the ADP-ribose level is nevertheless important compared to the time 0’ for all the groups in the different times of ischemia (p<0.01). Statistically significant differences are not considerable between the 31°C and the 34°C group.
Discussion
From the analysis of the results it can be suggested that in conditions of global ischemia and normothermia there is a progressive reduction of the main energetic metabolites. The fall in the ATP levels is surely not surprising. Nevertheless, many works in the literature refer to a rapid depletion of the energetic metabolite concentrations which would happen in the span of a few minutes from the beginning of the ischemia and would provoke the reduction of about 90% of the cellular reserves [14-18]. Such a net reduction is not at all documented in this study. The ATP in fact seems to be reducing gradually as time passes from the ischemic event, but even after 2 hours a certain value of energetic metabolites still available remains (Figure 1). The absence of a real fall of the ATP levels coincides with the impossibility of pointing out a cut-off value in the graph, beyond which the energetic metabolism could be irreparably compromised and the tissue irreversibly damaged. It is obvious that the considered temporal window frame is anyhow limited and it is therefore legitimate to expect that, after a longer ischemia time, a final depletion of the energetic cellular reserves will present itself; nevertheless, after two hours of global ischemia, it remains surprising to still observe the persistence of a significant quota of ATP. If the reduction of the ATP levels is confronted at the different temperatures examined in the research, it can be observed that the reduction of the cerebral temperature doesn’t seem able to preserve the energetic metabolism. Actually, after an initial slowdown of the ATP depletion, observable in the hypothermic rats after 30 minutes of ischemia, the following reduction of the energetic levels doesn’t differ in a substantial way among the three groups. In other parts of this dissertation it was discussed about the fact that the supposed capacity of the hypothermia to induce, in ischemia conditions, a slowdown of the fall of the ATP values, probable consequence of a reduction of the demand for oxygen and of a global inhibition of the cerebral metabolism, is non a unanimously recognized data [19]. Some authors in fact did not observe any modification in the ATP levels in conditions of hypothermia compared to the normothermic controls after situations of ischemia. So, as it was documented an initial protective effect induced by the temperature reduction after 30 minutes from the beginning of the ischemia it appears that by widening the temporal window frame of observation any protective role linked to the hypothermia seems irremediably lost. The data emerged from our research seems therefore in line to what stated by earlier experimental evidences in which it was observed a preservation effect of the energetic metabolism from the reduction of the cerebral temperature only in case of short length ischemic distress [20]. After 60 minutes of ischemia in our experimental model, the group of animals at 31°C seems to tend towards a higher conservation of the energetic levels, differentiating in a statistically significant manner from the other examined groups. This data could suggest that only the more marked reductions of the cerebral temperature would allow the preservation in time of that initial protective effect.
The increase, in conditions of ischemia, of the concentration of the final products of the catabolism of ATP, and in particular of the oxypurines (xanthine, hypoxanthine and uric acid), was confirmed in our experimental model. The higher availability of these compounds is at the base of the stimulation of the biochemical way that leads to the formation of free radicals by the xanthine-oxidase enzyme (XO), through the reaction reported below [21]:

The results of the oxidation reaction are represented by the superoxide (O2‾•) and by the peroxide of hydrogen or oxygenated water (H2O2). Another enzyme exists in the cerebral cellules, the Xanthine-Dehydrogenase (XD), that is equally responsible for the of the hypoxanthine’s metabolism; in this case the reaction of dehydrogenization doesn’t lead to the production of free radicals. The XO derives from the proteolytic clivage of the XD which is facilitated, in conditions of ischemia, by the presence of high concentrations of calcium which activate the proteases responsible for the transformation; in such a way the production of free radicals is consequently favored. It is important to note that, as it is inferred by the reported reaction, the formation of free radicals is a process that requires the availability of oxygen. In the present experimental model of global ischemia such availability is a function of a certain quantity of oxygen leftover at the tissue level and of the quota that the same tissue can acquire from the atmosphere.
This allows, albeit more controlled, for the production of those
toxic substances. In our opinion, this is a particularly important
concept because it reaffirms what is many times stated in other
parts of this dissertation: the production of free radicals is a
process typical of the reperfusion period that is nevertheless in
part influenced by mechanisms that begin to activate themselves
before the restoration of the hematic flux. Also in vivo it is actually
possible that one quota of leftover oxygen is available at the cellular
level; furthermore, the data correlates perfectly with the models of
temporary focal ischemia, in which the oxygen can spread through
diffusion towards the infarct areas from the adjacent zones still
perfused. In such conditions an increase of the formation of free
radicals happens, even before the occlusion at the base of the
situation of ischemia is removed [22,23]. Among the different ways
of formation of the oxygen derivate, the one that plays an important
role in the phase of ischemia in the strict sense is the reaction
mediated by the XO, while the others activate only at a later stage
when the hematic flux is reestablished [24,25].
As evidence of what has been stated, in the present model we
observe a progressive raise of the concentrations of uric acid which
represents the final product of the reaction previously described.
Considering then the trend of the MDA levels (Figure 2), expression
of the action of the free radicals on the membrane phospholipids, in
conditions of normothermia, it can be noted that the increase of the
concentrations, while present and clear, is not nearly comparable
to the burst of activity that the oxygen derivate normally show at
the moment of reperfusion, further evidence of the fact that only
a minimal quantity of these substances was able to be formed.
The most significant data is the one that differs from the analysis
of the MDA in hypothermia conditions. Both at 34 and 31ºC, at a
distance of 30 and 60 minutes from the beginning of the ischemia,
significant modifications of the MDA concentrations do not stand
out, if compared with the ones reached for the equal observation
times at 37ºC. After 2 hours of ischemia the group of animals at
31ºC is the only one to present a lower increment of MDA levels
compared to the rats at 37ºC.
These data do not contradict the by now confirmed capacity
of the reduction of cerebral temperature to reduce the formation
of free radicals in conditions of ischemia, but simply implicitly
imply a different mechanism of action. The hypothermia is in fact
essentially able to contrast the activity of the NOS and most of all of
the inducible isoform, which is mainly contained at the microglial
cellular level, which, in turn, are activated by the inflammation and
secondary damage processes that are carried out in the phase of
reperfusion. From the research of the Hyung Soo Han group [24,25]
previously described, it has emerged that the maximum peak of
formation of the two isoforms of the NOS shows up at the distance
of 24 hours from the ischemic event. It is true that from that
research it has also emerged that the intraischemic hypothermia
(33ºC) was able to reduce the activity and cellular expression of the
iNOS also at a distance of only 2 hours from the beginning of the
ischemic event, but is also true that the model used in that case was
one of a temporary focal ischemia and that the sample taken after 2
hours coincided with the restoring of the hematic flux. In a moment
of permanent global ischemia obtained through decapitation it is
unlikely to imagine the possibility that a situation of secondary
inflammation might occur; the only way of formation of free
radicals remains then the one described. In such conditions even
a moderate hypothermia (31ºC) results ineffective in contrasting
the formation of dangerous derivate of oxygen, since it is not able
to intervene in a significant manner on the reaction catalyzed by
the XO.
In this experimental model, in conditions of normothermia,
a progressive reduction of the concentrations of the nicotinic
coenzymes is observed. This phenomenon is expression of the
mitochondrial damage that happens following the ischemic
event: the minor availability of the NAD and NADP, that are
important electron acceptors, corresponds at a gradual block of the
mitochondrial respiratory chain and consequently of the oxidative
phosphorylation [26]. Such a process seems to be the result of
the previously described mitochondrial alterations that occur in
the first hours after the ischemic insult and that see involved, as a
main responsible, the intracellular calcium [27]. Nevertheless, from
the analysis of the results of this research emerges also another
data that could be correlated to the reduction of the levels of the
nicotinic coenzymes: the progressive increase of the quota of ADPribose.
The ribosylation of the ADP following the ischemic event
is a process mediated almost exclusively by the action of the PARP
system [28,29].
These cellular enzymes are activated through only one known
system, represented by the single filament breaking of the DNA
chain [30]. The alterations on account of the cellular nucleic acids
are the result of the free radical actions: the peroxynitrite first
and foremost, but also the hydroxyl radical and the superoxide
are the compounds mainly involved [31]. For that reason, both the
alterations at the DNA level both the subsequent activation of the
PARP system are generally phenomena typical of situations of focal
and temporary ischemia, showing up at the moment of the restoring
of the hematic flux and of the supply of oxygen. Nevertheless, in our
experimental model the presence of a certain quota of activity by
the free radicals is indisputable, as shown by the increase, even if
contained, of the MDA values. There are no reasons to thing that, in
the same manner in which a damage action is carried out toward the
phospholipidic membranes, an alteration could not be carried out,
mediated by the radicals, at cellular DNA level, able to activate the
enzymatic system of the poli-ADP-ribose-polymerase. This theory, without an objective evidence of the presence of DNA alterations
and without the use of specific inhibitors for the enzymatic systems
in question, cannot be considered completely well grounded.
Nevertheless, the registered raise of the ADP-ribose levels would
seem to support it. If so should be, the progressive reduction of the
NAD concentrations could be interpreted also in light of a direct
action by the PARP system on the nicotinic coenzyme, since these
enzymes are able to provoke a cleavage of the NAD, becoming the
main responsible of its depletion in conditions of focal ischemia
[29].
Furthermore, this model would demonstrate that an activation
of the PARP system, maybe even partial, can happen even while in
absence of a restoring of the hematic flux. Future research would
anyway be necessary to confirm this interesting theory. The
analysis of the trend of the ADP-ribose values and of the nicotinic
coenzymes in conditions of hypothermia provides important
elements of reflection. It can be observed that, both at 31 and a t
34ºC after 2 hours of ischemia a net fall of the ADP-ribose values
associated to the keeping, in the case of the group of rats at 31ºC,
or higher concentrations of NAD compared to the normothermic
controls. This data could indicate a protective effect induced by
the reduction of the cerebral temperature that would be exercised
directly towards the activity of the PARP system. As a matter of
fact, since significant modifications of the free radicals activity
between the groups of hypothermic animals and the one at 37ºC
were not recorded in the present experimental model, the reduced
concentrations of ADP-ribose documentable at 31 and also at 34ºC,
cannot be interpreted on the base of a minor activation of the
enzymatic system subsequent to a lower damaging action at the
nuclear level from the oxygen derivate; it is therefore more likely
that the hypothermia is capable to directly antagonize the action of
the enzymes, once that these are normally activated.
It is fair to underline that at 34ºC, together with a reduction
of the ADP-ribose concentrations mainly comparable to the one
that happens at 31ºC, a significant difference of the NAD levels
compared to those recorded at 37ºC is not evident; the data
should not come as a surprise, since it is to be remembered that
in conditions of ischemia other mechanism exist that can provoke
the reduction of the NAD levels, different from the cleavage action
exercised by the PARP system. It is sufficient to think that the
synthesis of the nicotinic coenzyme is an ATP dependent process
and that it is carried out partly inside the mitochondria that begin
to present marked alterations of their structure and function
already after an hour of the beginning of the ischemia [32,33].
Moreover, as it has been demonstrated by other researches [7,34],
the NADH molecule, just like the NADPH one, are another preferred
target of the hydroxyl radicals. The interaction of the OH• with
the NADH and NADPH provokes both their oxidation at NAD+ and
NADP+ and the production, at their expense, of ADP-ribose and
nicotinamide. This reaction would justify on one hand the drop
of the NAD levels, and on the other the increase of the ADP-ribose
concentrations. Moreover, it is important to underline that the
action of the hydroxyl radicals and the subsequent depletion of the
pool of nicotinic coenzymes jeopardize the correct functioning both
of the cellular energetic metabolism and of the oxide-reductive one,
making more complicated the recovery of the cellule in the possible
following phase of reperfusion.
Conclusion
Hypothermia is indeed one of the most important neuroprotective measures and through this animal model it was possible to demonstrate how even after 2 hours from an ischemic event a certain value of energetic metabolites still available remains. In addiction it seems that this protective effect might be due to the activity of the PARP system. Finally, metabolism interruption seems to be not an instantaneous process but rather something occurring in a longer period of at least 2 hours.
Acknowledgment
None.
Declarations of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
References
- Medvedeva YV, Ji SG, Yin HZ, Weiss JH (2017) Differential Vulnerability of CA1 versus CA3 Pyramidal Neurons After Ischemia: Possible Relationship to Sources of Zn2+ Accumulation and Its Entry into and Prolonged Effects on Mitochondria. J Neurosci Off J Soc Neurosci 37: 726-737.
- Ahrendsen JT, Grewal HS, Hickey SP, Culp CM, Gould EA, et al. (2016) Juvenile striatal white matter is resistant to ischemia-induced damage. Glia 64: 1972-1986.
- Bartsch T, Döhring J, Reuter S, Finke C, Rohr A, et al. (2015) Selective neuronal vulnerability of human hippocampal CA1 neurons: Lesion evolution, temporal course, and pattern of hippocampal damage in diffusion-weighted MR imaging. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 35: 1836-1845.
- de la Torre JC, Saunders J, Fortin T, Butler K, Richard M (1991) Return of ATP/PCr and EEG after 75 min of global brain ischemia. Brain Res 542(1): 71-76.
- Hossmann KA, Grosse Ophoff B (1986) Recovery of monkey brain after prolonged ischemia. I. Electrophysiology and brain electrolytes. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 6: 15-21.
- Derbyshire DR, Clark RG (1980) Cerebral recovery after prolonged global brain ischaemia. Lancet Lond Engl 2: 637.
- Choi D-H, Kim J-H, Lee K-H, Kim H-Y, Kim Y-S, et al. (2015) Role of neuronal NADPH oxidase 1 in the peri-infarct regions after stroke. PloS One 10: e0116814.
- Karnatovskaia LV, Wartenberg KE, Freeman WD (2014) Therapeutic hypothermia for neuroprotection: history, mechanisms, risks, and clinical applications. The Neurohospitalist 4: 153-163.
- Drury PP, Gunn ER, Bennet L, Gunn AJ (2014) Mechanisms of hypothermic neuroprotection. Clin Perinatol 41(1): 161-175.
- Zhang Z, Zhang L, Ding Y, Han Z, Ji X (2018) Effects of Therapeutic Hypothermia Combined with Other Neuroprotective Strategies on Ischemic Stroke: Review of Evidence. Aging Dis 9(3): 507-522.
- Park CK, Jun SS, Kim MC, Kang JK (1998) Effects of systemic hypothermia and selective brain cooling on ischemic brain damage and swelling. Acta Neurochir Suppl 71: 225-228.
- Busto R, Dietrich WD, Globus MY, Valdés I, Scheinberg P, et al. (1987) Small differences in intraischemic brain temperature critically determine the extent of ischemic neuronal injury. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 7: 729-738.
- Horn M, Schlote W, Henrich HA (1991) Global cerebral ischemia and subsequent selective hypothermia. A neuropathological and morphometrical study on ischemic neuronal damage in cat. Acta Neuropathol (Berl) 81(4): 443-449.
- Pimentel VC, Zanini D, Cardoso AM, Schmatz R, Bagatini MD, et al. (2013) Hypoxia-ischemia alters nucleotide and nucleoside catabolism and Na+,K+-ATPase activity in the cerebral cortex of newborn rats. Neurochem Res 38(4): 886-894.
- Melani A, Corti F, Stephan H, Müller CE, Donati C, et al. (2012) Ecto-ATPase inhibition: ATP and adenosine release under physiological and ischemic in vivo conditions in the rat striatum. Exp Neurol 233(1): 193-204.
- Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79(4): 1431-1568.
- Lipton P, Whittingham TS (1982) Reduced ATP concentration as a basis for synaptic transmission failure during hypoxia in the in vitro guinea-pig hippocampus. J Physiol 325: 51-65.
- Martin RL, Lloyd HG, Cowan AI (1994) The early events of oxygen and glucose deprivation: Setting the scene for neuronal death? Trends Neurosci 17(6): 251-257.
- Nilsson L, Kogure K, Busto R (1975) Effects of hypothermia and hyperthermia on brain energy metabolism. Acta Anaesthesiol Scand 19(3): 199-205.
- Welsh FA, Sims RE, Harris VA (1990) Mild hypothermia prevents ischemic injury in gerbil hippocampus. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 10: 557-563.
- Hagberg H, Andersson P, Lacarewicz J, Jacobson I, Butcher S, et al. (1987) Extracellular adenosine, inosine, hypoxanthine, and xanthine in relation to tissue nucleotides and purines in rat striatum during transient ischemia. J Neurochem 49(1): 227-231.
- Barone FC, Feuerstein GZ (1999) Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 19: 819-834.
- Sun M-S, Jin H, Sun X, Huang S, Zhang F-L, et al. (2018) Free Radical Damage in Ischemia-Reperfusion Injury: An Obstacle in Acute Ischemic Stroke after Revascularization Therapy. Oxid Med Cell Longev 2018: 3804979.
- Liu XH, Kato H, Nakata N, Kogure K, Kato K (1993) An immunohistochemical study of copper/zinc superoxide dismutase and manganese superoxide dismutase in rat hippocampus after transient cerebral ischemia. Brain Res 625(1): 29-37.
- Panahian N, Yoshida T, Huang PL, Hedley-Whyte ET, Dalkara T, et al. (1996) Attenuated hippocampal damage after global cerebral ischemia in mice mutant in neuronal nitric oxide synthase. Neuroscience 72(2): 343-354.
- Kroemer G, Dallaporta B, Resche-Rigon M (1998) The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol 60: 619-642.
- Dubinsky JM, Levi Y (1998) Calcium-induced activation of the mitochondrial permeability transition in hippocampal neurons. J Neurosci Res 53(6): 728-741.
- Lo EH, Bosque-Hamilton P, Meng W (1998) Inhibition of poly (ADP-ribose) polymerase: Reduction of ischemic injury and attenuation of N-methyl-D-aspartate-induced neurotransmitter dysregulation. Stroke 29(4): 830-836.
- Takahashi K, Greenberg JH, Jackson P, Maclin K, Zhang J (1997) Neuroprotective effects of inhibiting poly (ADP-ribose) synthetase on focal cerebral ischemia in rats. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 17: 1137-1142.
- Berger NA (1985) Poly (ADP-ribose) in the cellular response to DNA damage. Radiat Res 101(1): 4-15.
- Chen J, Jin K, Chen M, Pei W, Kawaguchi K, et al. (1997) Early detection of DNA strand breaks in the brain after transient focal ischemia: Implications for the role of DNA damage in apoptosis and neuronal cell death. J Neurochem 69(1): 232-245.
- Petito CK, Pulsinelli WA (1984) Sequential development of reversible and irreversible neuronal damage following cerebral ischemia. J Neuropathol Exp Neurol 43(2): 141-153.
- Yamamoto K, Hayakawa T, Mogami H, Akai F, Yanagihara T (1990) Ultrastructural investigation of the CA1 region of the hippocampus after transient cerebral ischemia in gerbils. Acta Neuropathol (Berl) 80(5): 487-492.
- Tavazzi B, Di Pierro D, Amorini AM, Fazzina G, Galvano M, et al. (2000) Direct NAD(P)H hydrolysis into ADP-ribose(P) and nicotinamide induced by reactive oxygen species: A new mechanism of oxygen radical toxicity. Free Radic Res 33(1): 1-12.
