 Case Report
Case ReportAbstract
Background: Cronkhite-Canada syndrome is a rare disease of unknown etiology. Until now, no evidence exists to suggest a familial predispositions.
Case Presentation: We present two familial cases: one a 31-years old female patient with chronic watery intermittent diarrhea and abdominal pain for 12 years and hematochezia for one month. She aslo had hyperpigmentation over hands, onchodystrophy, alopica, hypogeusia, anorexia and weight loss (since 6 years). Her 9-years old son also experienced intermittent abdominal pain, diarrhea and hematochezia for 1 year. Gastrointestinal endoscopy showed multiple polyps in stomach and colon in both cases. Suspecting a genetic association, we assessed the sequence variation of the exon region of Polyposis related gene in the mother using next generation sequencing (NGS). A mutation of APC gene c.3921-3925delAAAAG (p.Ile1307fsX6) was found. This mutation site was examined in the patient’s mother, father, three siblings and son by PCR-sanger sequencing. Only the son had a mutation in the same site. The familial case of Cronkhite-Canada syndrome was suspected.
Conclusion: So far, there is no strong evidence to suggest a familial predisposition. We report a case of Cronkhite-Canada syndrome in mother and son suggestive of familial predispositions. We have found that they have same mutations in APC gene by genetic testing.
Keywords: Cronkhite-Canada Syndrome, Familial Case, Gastrointestinal Polyposis
Abbreviations: CT: Computed Tomography; CCS: Cronkhite-Canada Syndrome: APC: Adenomatous Polyposis Coli
Introduction
Cronkhite-Canada syndrome is a rare disease presented with multiple gastrointestinal polyps, alopecia, onchodystrophy, skin hyperpigmentation, weight loss, diarrhea and intermittent abdominal pain. It was first described in 1950 where more than 550 cases has been reported world-wide with more than 75% who are originated from Japan [1-3]. The etiology of the disease is undetermined, and the no familial predisposition has been reported [4,5]. To our knowledge, only one familial case of Indian decent have been reported [5] but no genetic testing has been done. Here, we report a case of Cronkhite-Canada syndrome in a Chinese mother and son.
Case Presentation
A 31-years old woman has visited our hospital with a history of chronic watery intermittent diarrhea and abdominal pain for 12 years and hematochezia for 1 month. She also had hyperpigmentation over hands, onchodystrophy, alopica, hypogeusia, anorexia and weight loss (for past 6 years). Family history of the patient revealed mother was suffering over 10 years from diarrhea without accepting medical treatment. Father suffered from cerebral infarction a year ago. All three siblings are in good health and they have no family history of polyposis. On physical examination patient’s vital signs were normal. Her examination was significant for diffuse alopecia (Figure 1A), hyperpigmentation over hands and onchodystrophy (Figure 1B). The abdominal examination was unremarkable and had no lymphadenopathy. There was no abnormality seen on CT scan of abdomen and MRI scan for bone and brain. The white blood cells count was 3.50*10^9/L; red blood cells 3.9*10^12/L; hemoglobin 115g/L; lymphocyte 39.9%; mononuclear cell 11.5%; neutrophil 1.6410^9/L↓; hematocrit 0.348L/L ↓; and no abnormal reticulated red cell count was identified. The electrolytes: magnesium 1.27mmol/L; globulins 36.8g/L; apolipoproteins A10.87g/L and the upper endoscopy revealed multiple sporadic small polyps in the stomach (Figure 2A).
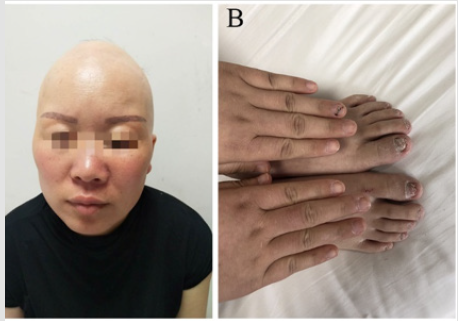
Figure 1: (A) Thirty-one years old female with alopecia over scalp.
(B) Dystrophic changes over toes nails and hyperpigmentation over hands.
Colonoscopy showed numerous sessile polyps scattered through the colon (Figure 2B). The polyps were ranged in size of 2mm to 3 cm in size. Ileal polyps were also present. Capsule endoscopy showed normal small intestine. She was also accompanied her 9year old son whose was suffering from intermittent abdominal pain, diarrhea and bloody stools. Gastrointestinal endoscopy showed multiple polyps in stomach, duodenum, cecum and colon (Figures 2C & 2D). Histopathology of the mother gastric polyps were hyperplasic and ilial polyps showed tubulo-villous adenomatous with low grade dysplasia (Figures 3A & 3B). Biopsy of the large colonic polyps showed tubulo-villous adenomas with low-grade dysplasia and inflammatory stroma (Figures 3C & 3D). Immunohistochemical staning showed IgG positive (Figure 3E) and IgG4 weekly positive (Figure 3F). Analysis of the sequence variation of the exon region of 18 genes associated with polyposis (APC, MLH1, MSH2, MSH6, PMS2, AXIN2, BMPR1A, EPCAM, MLH3, MUTYH, PMS1, PTEN, SMAD4, STK11) in the mother by next generation sequencing (NGS). Genetic analysis revealed mutation of APC gene c.3921- 3925delAAAAG (p.Ile1307fsX6). We then analyzed the mutation site of the patient, her mother, father, three siblings and her son by PCR-sanger sequencing. Mutation of the same site was only detected in her son.
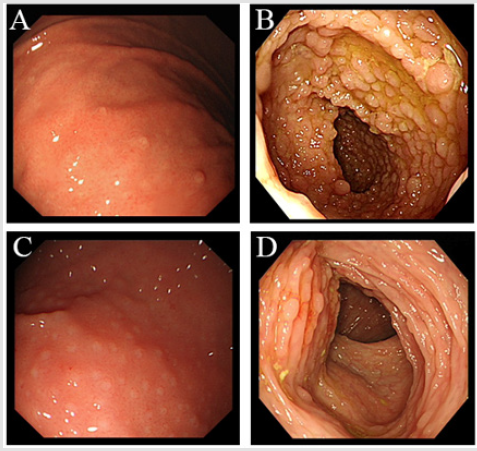
Figure 2: (Endoscopic view of the 31 years old patient
(A) Sporadic small polyps in the stomach; (B) Colon Endoscopic view of the 9 years old son; (C) Polyps in the stomach;
(D) colon.
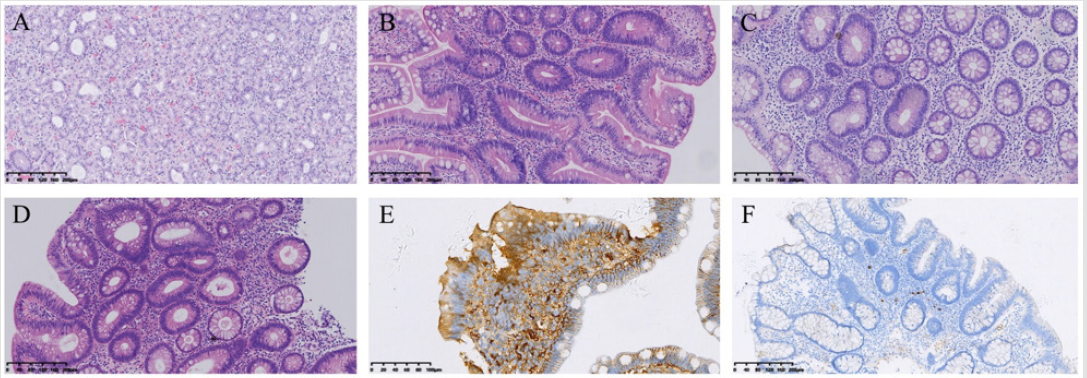
Figure 3: Histopathology findings of
(A,B) stomach and ileum polyps.
(C,D) Colon.
(E,F) Immunohistochemical staning of intestine.
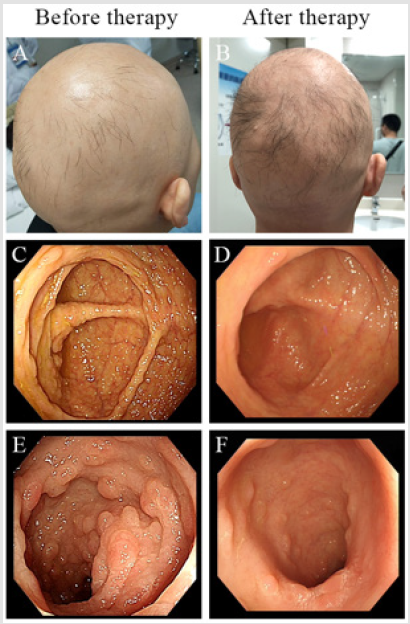
Figure 4: Endoscopy view before and after the therapy.
The site c.3921-3925delAAAAG (p.Ile1307fsX6) as deletion mutation has been reported in patients with hereditary gastrointestinal polyposis, which is a pathogenic mutation site, which can lead to adenoma, autosomal dominant diseases such as polyposis, brain tumor-polyposis syndrome type 2, gardner syndrome [6-10]. The mother was initially started on total parenteral nutrition to provide rest to the bowel and then slowly started on enteral feeds along with methylpredinsolone (40 mg orally once a day). During follow-up endoscopy showing reduction in number and size of the polyps (Figure 4). Considering the son is too young to tolerate the side effects methyprednisolone, mesalazine enteric-coated tablets was given (0.25g orally t.i.d). The symptoms of diarrhea, hematochezia and abdominal pain released but polyps burden did not reduce significantly. The Nutritional condition of body improved.
Discussion and Conclusion
In 1955 Cronkhite and Canda described regarding the syndrome of multiple gastrointestinal polyps, oncholysis, alopecia, skin hyperpigmentation, diarrhea and abdominal pain [1-3]. Since then, nearly 550 cases of Cronkhite-Canada syndrome have been reported worldwide. The average age of onset is estimated to be between 50 - 60 years with a 3:2 male/female ratio [4,5]. Considering the present scenario, the etiology of the Cronkhite- Canada syndrome remains unclear. However, an autoimmune process is suspected due to increase of IgG4 levels which are found in Cronkhite-Canada syndrome polyps [4-6]. Till date, no evidence exists to suggest a familial predisposition. The possibilities of symptomatic offspring or afflicted patients were not excluded. Here, we got a case of Cronkhite-Canada syndrome in a 31-year-old mother and 9-years old son suggestive of familial predispositions. We have found that they have same mutations in APC gene by genetic testing. The optimal treatment of Cronkhite-Canada syndrome is currently unknown due its rarity and unclear etiology. Nutritional support aimed for improving electrolytes and mineral deficiencies that can rarely induce complete remission as observed in our patients. In fact, current studies favor a combination therapy based on nutritional support and corticosteroids. Unfortunately, due to rarity of this disease, optimal screening protocol has also not been developed, although annual endoscopic surveillance has been widely practiced.
Despite therapy, mortality rate approaches 65%. However, there are long term survivors and some individuals who undergo spontaneous remission. There is no effective therapy for this condition, despite the use of systemic anabolic steroids has been reported. Resection of stomach and colorectum maybe indicated if heavily carpeted with polyps or source of excessive bleeding, bowel obstruction or cancer. In conclusion, based on genetic testing we concluded that, mother may have developed CCS from FAP, there is a possibility that the son may also develop CCS later because we found the mutation in the sane gene. The average age of CCS is 30-80 years. The symptoms in the son are not obvious as he is too young. Our results have significance scope for developing an optimal screening and treatment for the patients who are suffering from Cronkhite-Canada syndrome.
Declarations
Acknowledgments
The authors thank Professor David Y. Graham, Professor of Medicine, Molecular Virology and Microbiology, Baylor College of Medicine for his encouragements and assistance in revising the manuscript of this article.
Funding
No funding was obtained for this study.
Author’s contribution
Study concept and design: Hanbing Ning
Acquisition of data: Jingyun Wang, Qiaoyu Jia, Hongtao Wen
Manuscript writing: Jingyun Wang. Qiaoyu Jia.
Administrative, technical or material support: Wenquan Lu,
Shuai Lv, Yingxia Li
Critical revision of manuscript: Hanbing Ning
Availability of data and materials
All data generated or analyzed during this study are included in this articles.
Consent for publication
Written informed consent of publication of their images and other information was obtained from the patient.
Competing interess
The authors declared that they have no competing interests.
Ethics approval and consent to participate
The present study was undertaken under the guidence of the ethics comittee of The First Affiliated Hospital of Zhengzhou University. The authors have declared that no ethical conflicts.
References
- Burke AP, Sobin LH (1989) The pathology of Cronkhite-Canada polyp. A comparison to juvenile polypposis. Am J Surg Pathol 13(11): 940-946.
- Bandyopadhyay D, Adrija Hajra, Vijayan Ganesan, Suvrendu Sankar Kar, Debarati Bhar, et al. (2016) Cronkhite-Canada syndrome: a rare cause of chronic diarrhoea in a young man. Case Rep Med 2016: 4210397.
- Rubio CA, Björk J (2016) Cronkhite-Canada syndrome - A Case report.Anticancer Res 36(8): 4215-4217.
- She Q, Jiang JX, Si XM, Tian XY, Shi RH, et al. (2013) A severe course of Cronkhite-Canada syndrome and the review of clinical features and therapy in 49 Chinese patients. Turk J Gastroenterol 24(3): 277-285.
- Patil V, Patil LS, Jakareddy R, Verma A, Gupta AB (2013) Cronkhite-Canada syndrome: a report of two familial cases. indian J Gastroenterol 32(2): 119-122.
- Baldino ME, Koth VS, Silva DN, Figueiredo MA, Salum FG, et al. (2019) Gardner syndrome with maxillofacial manifestation: A case report. Spec Care Dentist 39(1): 65-71.
- Half E, Dani B, Paul R (2009) Familial adenomatous polyposis. Orphanet J Rare Dis 4: 22.
- Sokic Milutinovic A (2019) Appropriate Management of Attenuated Familial Adenomatous Polyposis: Report of a Case and Review of the Literature. Dig Dis 37(5): 400-405.
- Kennedy RD, Potter DD, Moir CR, El Youssef M (2014) The natural history of familial adenomatous polyposis syndrome: a 24-year review of a single center experience in screening, diagnosis, and outcomes. J Pediatr Surg 49(1): 82-86.
- Yun SH, Cho JW, Kim JW, Kim JK, Park MS, et al. (2013) Cronkhite-Canada syndrome associated with serrated adenoma and malignant polyp: a case report and a literature review of 13 Cronkhite-Canada syndrome cases in Korea. Clin Endosc 46(3): 301-305.
