 Research Article
Research ArticleAbstract
The loss of dystrophin expression leads to Duchenne muscular dystrophy (DMD), the most common form of muscular dystrophies. This genetic disorder induces muscle damage and significant secondary inflammatory injury, with muscle inflammatory foci mostly composed of macrophages and few T lymphocytes. We previously studied the scarcity of lymphoid cells in mdx mice muscles, a murine model for DMD, and published that a subpopulation of blood T lymphocyte sheds the CD62L molecule from the cell membrane. This shedding is induced by P2X7 activity and leads to reduced T lymphocyte endothelial adhesion in vitro. After P2X7 impairment in vivo, these T lymphocytes retained the selectin, homed to the cardiac muscle, and increased muscle damage. However, we did not define if the increased population of CD62L- T lymphocyte was composed of naϊve or antigen-primed cells and decided to revisit this subject. We observed that the altered phenotype is not restricted to the CD62L, as long as CD127 and CD44 are also downregulated in a P2X7-dependent manner. CD44 is a lymphocytehoming receptor that binds mainly to hyaluronan and regulates processes such as cell adhesion, angiogenesis, inflammation, cellular activation, cytoskeleton rearrangements, and others. The present results confirm and extend our previous publication and we attribute the reduced capacity of T lymphocyte homing to non-lymphoid tissues in mdx mice to a broader panel of membrane molecules affected by P2X7 activity.
Keywords: Duchenne Muscular Dystrophy; T Lymphocyte Migration; Adhesion Molecules; Inflammation; Cd44
Abbreviations: DMD: Duchenne Muscular Dystrophy, DGC: Dystrophin Glycoprotein Complex, PDGFR: Platelet-Derived Growth Factor, EGFR: Epidermal Growth Factor, HA: Hyaluronan
Introduction
Duchenne muscular dystrophy (DMD) is the most common form of muscular dystrophies, affecting 1 in 3,600 to 6,000 male births. This neuromuscular disorder is caused by the absence of dystrophin [1], a protein that links intracellular F-actin to the dystrophin glycoprotein complex (DGC) [2], a multiprotein structure that connects the muscle fibers to the extracellular matrix. The lack of dystrophin destabilizes the muscle fibers during contraction cycles, leading to progressive cell damage through membrane leakage. The first symptoms appear in childhood, when the patients have difficulties in keeping up with peers and experience progressive weakness, followed by loss of ambulation early in the second decade of life. Because of the progressive myonecrosis, inflammatory cells invade skeletal and cardiac muscles and promote secondary pathological damage [3]. Macrophages [4] are the predominant cell type found in inflammatory foci in DMD and it was observed that these cells can be cytotoxic against myofibers in vitro. Moreover, macrophage depletion leads to reduced myonecrosis, indicating macrophage-dependent cytotoxicity [5].
Regarding αβ T lymphocyte, their role has also been mostly associated with increased muscle damage, as the depletion of CD4+or CD8+ T cells in young mdx mice, a murine model for DMD, led to reduced skeletal muscle damage. Besides, the adoptive transfer of mdx splenocytes combined with muscle extract treatment induced CD8+ T lymphocyte migration to muscles and injury [6]. This result may be due to the release of perforin, as null mutants for both dystrophin and perforin had less muscle pathology [7]. On the other hand, we published that γδ T lymphocyte migrate to the heart of twelve-week old mdx mice and they are protective of the muscle; once in vivo depletion of these cells increased muscle damage [8]. In general, leukocyte interaction with endothelial cells is mainly mediated by CD62L, followed by activated integrins engagement. In the particular case of T lymphocyte, the required molecular repertoire for transmigration and the importance of CD62L were controversial for a long time, partially because of the complex division of cellular subpopulations that follows antigen exposure. Murine antigen-primed and naïve T lymphocyte subpopulations can now be defined based on the expression of two main categories of molecules, which are homing receptors that allow the migration to secondary lymphoid organs (CD62L) or non-lymphoid tissues (CD44 and CD197), and interleukin-7 receptor alpha chain (CD127) that is required for naïve and memory cells long-term survival [9].
Currently, it is believed that the CD62L is not required for effector T lymphocyte migration to inflamed tissues, and these cells are defined as CD62L-CD44highCD197-CD127-. Moreover, effector memory T lymphocyte also lose the expression of CD62L, being recognized as CD62L-CD44highCD197-CD127+. On the other hand, central memory (CD62L+CD44highCD197+CD127+) and naïve T lymphocyte (CD62L+CD44+CD197+CD127+) express the selectin and depend on this phenotype for cell function [10]. We previously studied if the reduced number of T lymphocyte in mdx mice muscles could be associated with an altered repertoire of endothelial adhesion molecules, which could compromise endothelial extravasation to damaged muscles. We observed no altered expression of LFA-1, CD43, or CD49d in blood T lymphocyte when comparing C57BL/10 with mdx mice, and there were normal levels of the CD62L expression in blood granulocytes and monocytes [11]. However, we observed in mdx mice blood an aberrant subpopulation of about 30% of T lymphocyte as CD62Ldue to P2X7-dependent membrane shedding [11]. We also found that these CD62L- T lymphocyte had a reduced adhesion capacity to endothelial cells in vitro.
Moreover, P2X7 inhibition in vivo restored normal levels of CD62L expression, increased T lymphocyte migration to the heart, and led to cardiac damage. Once mdx mice show neither immune depression nor increased susceptibility to pathogens, we used the CD69 marker to evaluated if the CD62L- subpopulation was composed of naïve or antigen-primed T lymphocyte, but the results were inconclusive [11]. Therefore, we decided to revisit our findings and the phenotypic profile of blood T lymphocyte in mdx mice. We confirmed the P2X7-dependent downregulation of CD62L and observed that these cells have an atypical phenotype also regarding CD44 and CD127 expression, as triple-negative cells, all downregulated by P2X7-activity. Therefore, this subpopulation does not fall in any of the previously described T lymphocyte phenotypes, being CD3+CD62L-CD44-CD127-. In regards to CD44, this is an important molecule for T lymphocyte function, binding mainly to Hyaluronan (HA) but also to osteopontin, collagens, membrane metalloproteinases, growth factors, and others [12]. There is the standard form of CD44 (CD44s) and several variant forms (CD44v) that result from tightly regulated alternative splicing and post-translational modifications, like glycosylation, all with high affinity for HA [12].
CD44 can also signal as a coreceptor in multifunction platforms associated with molecules such as the receptors of Vascular Endothelial Growth Factor (VEGFR), Epidermal Growth Factor (EGFR), and TGFβ (TGFβR), leading to the production of proinflammatory cytokines, proliferation, apoptosis protection, tissue invasion, angiogenesis, and cytoskeleton rearrangement [13]. The membrane engagement of CD44 with the receptor of Platelet- Derived Growth Factor (PDGFR) seems to block signal transduction through CD44 binding and the positive signaling pathways are based on PI3K/Akt and the engagement of glycogen synthase kinase 3 beta (GSK3β) and β-catenin; Erk; and Ras, for example. CD44 signaling also plays an essential role in the rolling and extravasation of lymphocytes to inflammatory foci [14], acting on at least three checkpoints, which are the binding to HA expressed by endothelial cells; the interaction of CD44v with members of the selectin family, and finally with VLA-4 for transendothelial migration [15]. Therefore, the CD44 downregulation on T lymphocyte membrane may affect cellular activation, adhesion, and function in inflammatory sites and, along with CD62L and CD127, may be responsible for the reduced transmigration of T lymphocyte to muscles in mdx mice.
Materials and Methods
Animals
All experiments were conducted using male twelve weekold mdx mice and age-matched C57BL/10 control mice obtained from the Center for Breeding of Laboratory Animals at Fundação Oswaldo Cruz. All mice were housed for seven to ten days before experimentation at the Center for Animal Experimentation under environmental factors and sanitation according to the Guide for the Care and Use of Laboratory Animals (8th edition, 2011). The Fiocruz Committee of Ethics in Research (L0006/2015) approved this project, according to resolution 196/96 of the National Health Council of Brazilian Ministry of Health.
Brilliant Blue G treatment
Brilliant Blue G (BBG) (Sigma-Aldrich, MO, USA) was diluted in vehicle solution (calcium- and magnesium-free PBS/0.2% dimethyl sulfoxide (Sigma-Aldrich)) to prepare a stock solution (3mg/mL). For in vivo treatment, mdx and C57BL/10 mice were weighed once a week during drug administration and received 100 μL of freshly prepared BBG (45.5 mg/kg) or vehicle via intraperitoneal injection at every 48 hours. The mice were treated from nine to twelve weeks of age.
Flow Cytometry
Peripheral blood mononuclear cells (PBMCs) were collected by cardiac puncture and the lymphocytes were obtained from the inguinal lymph nodes by mechanical maceration. All cells were processed as published [11] and incubated with previously titrated anti CD3-PECF 594, anti CD127-PE-Cy7, anti CD44-Alexa Fluor 488 (clone IM7, reactive to all isoforms of CD44), anti CD62L-PE, anti CD69-APC (all obtained from Biolegend, CA, USA). All samples were acquired in a FACSAria II (BD Biosciences, CA, USA) and data analysis was performed using FloJo version 10 (Beckman Coulter, CA, USA). The doublets exclusion was done in FSC Area x FSC Height dot plots and the lymphocyte gate was defined using CD3 labeling.
Dot Blotting
Blood samples were obtained from mdx or C57BL/10 mice by cardiac puncture in the absence of heparin; the samples were then left at room temperature for thirty minutes and processed as published [11]. Briefly, protein quantification was performed using a Pierce BCA protein assay kit (Thermo Fisher, MA, USA), and the amount of proteins indicated in the figure legends was transferred in triplicates to nitrocellulose membranes. The samples were then incubated with anti-CD62L or anti-CD44 (Biolegend) for one hour at room temperature, washed twice inTris buffer, and incubated for one hour with goat anti-rat HRP-conjugated secondary antibody (Vector Laboratories, CA, USA). The primary antibody was suppressed for negative controls. The revelation was performed using DAB (3, 3 –diaminobenzidine) solution (Vector Laboratories) and densitometry units were obtained using Image Studio Lite software version 4.0 (Licor Biosciences, NE, USA).
Statistical Analysis
All data were expressed as arithmetic mean ± SD. Before the statistical analysis, we used the Shapiro-Wilk test to confirm if the samples had a normal distribution (R Core Team (2019). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.Rproject. org/.). Then we used two-way ANOVA with Bonferroni posttest (GraphPad Prism version 4.00 software; GraphPad Software, CA, USA) and the adopted significance level was α = 5 % (P<0.05) for all statistical analyses.
Results
We previously published that a subpopulation of about 30% of blood T lymphocyte collected from twelve-week old mdx mice is CD3+CD62L-, different from age-matched control C57BL/10 and six-week old mdx mice that had less than 15% in average [11]. Moreover, we observed that the CD62L downregulation was mediated by P2X7 activity and postulated that the lack of CD62L would lead to insufficient endothelial adhesion and reduced transmigration of possibly early-activated effector T lymphocyte. However, the phenotypic analysis of this subpopulation was not conclusive and we could not categorize these cells as naïve or antigen-primed cells. This is a central point; as effector and effector memory T lymphocyte are CD62L- and, therefore, these cells are not expected to be targeted for selectin shedding. On the other hand, naïve and specially central memory T lymphocyte express CD62L, and they could be shedding the selectin. We then decided to revisit our data using an extended panel of T lymphocyte markers aiming to understand T lymphocyte phenotypic modulations in mdx mice and its impact on cellular migration to muscles. We isolated PBMC from twelve-week old mdx and C57BL/10 mice treated or not with BBG (a P2X7 pharmacological blocker) and observed that P2X7 activity modulates additional molecules on T lymphocyte membrane.
Using a different phenotypic panel and analysis strategy that was used previously [11], we observed in control C57BL/10 mice a minor T lymphocyte subpopulation of about 3% (in the morphological gate of lymphocytes and CD3+ cells) that was CD62LCD44- (Figure 1a) and about 83% of these cells were CD69- CD127+ (Fig 1A). Moreover, when we treated C57BL/10 mice with BBG in vivo, there were no important variations in this phenotype (Figure 1b). However, in mdx mice, 15 to 20% of the CD3+ population was CD62L- CD44- and there was a downregulation of CD127 in these cells (Figure 1c), with only about 25% of the cells as CD127+. Moreover, most CD62L- CD44- cells were CD69- (Figure 1c) and there was a subpopulation of about 12% as CD127- CD69+ cells (Figure 1c). The in vivo treatment of mdx mice with BBG reduced the CD62L- CD44- population (Figure1D), restoring the profile observed in C57BL/10 control animals (Figures 1a and 1b). These results show that CD62L, CD44, and (in most cells) CD127 are all downregulated by P2X7 activity. Once CD62L is a central molecule for naïve T lymphocyte homing to lymph nodes, where antigen interaction takes place for cellular activation, we evaluated the phenotype of T lymphocyte in inguinal lymph nodes of twelve-week old C57BL/10 and mdx mice. We observed no differences in lymph nodes cellularity when comparing both mouse lineages (data not shown), and there was a minor population of CD62L- CD44- T lymphocyte that was mostly CD69- CD127+ (Figure 2a), similar to what was observed in blood of control mice (Figure 1a). There were no modulations in this subpopulation after BBG treatment (Figure 2b) and a similar phenotypic profile was observed in untreated (Figure 2c) and BBG-treated (Figure 2d) mdx mice. These results confirm previous data in the literature that indicate no alterations in lymph nodes structure or lymphoid migration in the absence of dystrophin.
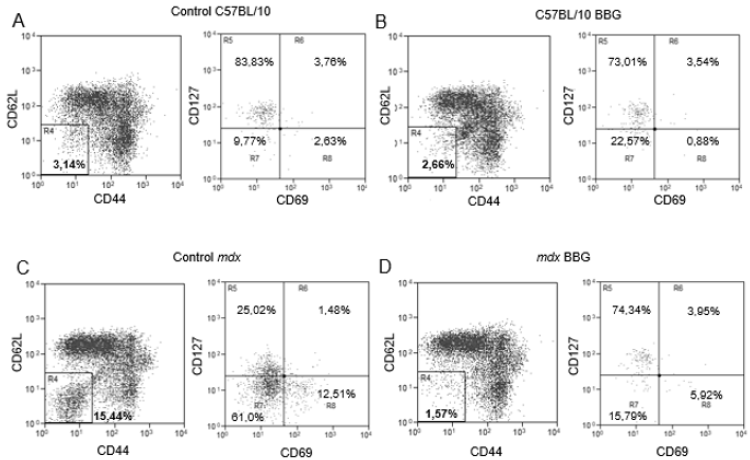
Figure 1: Phenotypic analysis of blood T lymphocytes. PBMC from twelve week-old C57BL/10 (A and B) or mdx mice (C and D), treated (B and D) or not (A and C) with BBG were labeled with anti-CD3, anti-CD69, anti-CD62L, anti-CD44, and anti-CD127 monoclonal antibodies for flow cytometry analysis. The analysis of CD62L and CD44 was done in the gates of lymphocytes and CD3+ cells, and the analysis of CD127 and CD69 was done in CD62L CD44 double negative T cells (R4). Subpopulations percentages are shown in each gate. Data are representative of three independent experiments with five to eight mice per group.
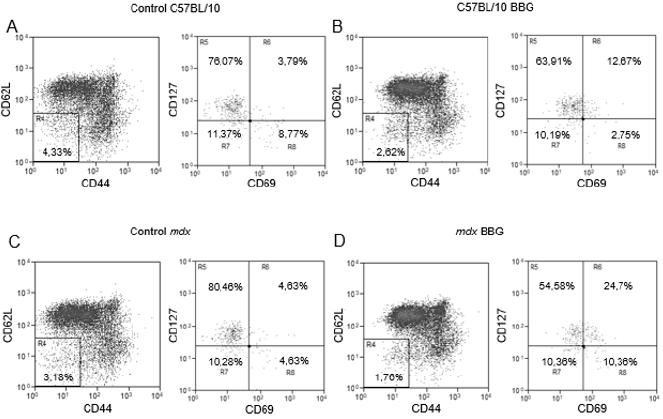
Figure 2: Phenotypic analysis of T lymphocytes from inguinal lymph nodes. Lymphocytes from twelve week-old C57BL/10 (A and B) or mdx (C and D) mice, treated (B and D) or not (A and C) with BBG were labeled with anti-CD3, anti-CD69, anti-CD62L, anti-CD44, and anti-CD127 monoclonal antibodies for flow cytometry analysis. The analysis of CD62L and CD44 was done in the gates of lymphocytes and CD3+ cells, and the analysis of CD127 and CD69 was done in CD62L CD44 double negative T cells (R4). Subpopulations percentages are shown in each gate. Data are representative of three independent experiments with five to eight mice per group.
As we previously observed that the CD62L was shed from blood T lymphocyte membrane and the concentration of soluble CD62L was higher in mdx mice serum [11], we evaluated if the CD44 was also shed from T cell membrane. This analysis was done by dot blotting in mice sera and we observed similar levels of this molecule in all groups studied (Figure 3a), suggesting no CD44 shedding from the cell membrane in mdx mice. Moreover, and as observed in our previous study, soluble CD62L was increased in mdx mice sera when compared with C57BL/10 counterparts (Figure 3b) and this shedding was better evidenced when we used 20 μg of total proteins. The BBG treatment reduced soluble levels of CD62L in mdx mice blood, not affecting the levels of this molecule in C57BL/10 mice (Figure 3b). Taken together, our data indicate a P2X7-dependent downmodulation of CD62L and CD44 but only CD62L is shed from T lymphocyte membrane.
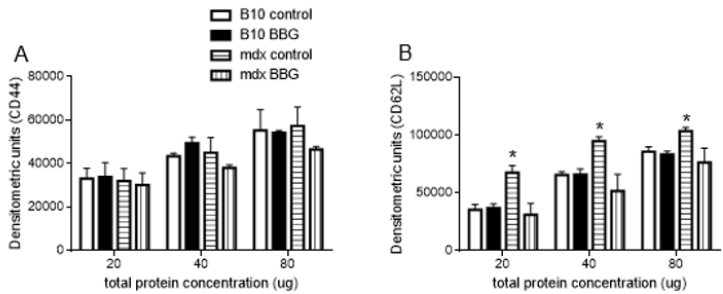
Figure 3: Blood soluble CD44 and CD62L in mdx and C57BL/10 mice. Serum concentrations of CD44 (A) or CD62L (B) from twelve-week-old mdx mice treated or not with BBG were evaluated by dot blotting using 20, 40, or 80 μg of total proteins per slot. The membranes were incubated with anti-CD44 or anti-CD62L, then with HRP-conjugated goat anti-rat secondary antibody and revealed using DAB. For negative control, the same amount of total proteins were immobilized, and the primary antibody was suppressed. Data were normalized by negative controls, and densitometric units are expressed as means +SD obtained from three independent experiments with five to eight mice per group. *P < 0.05.
Discussion
In many inflammatory muscle diseases, such as most inflammatory myopathies and T. cruzi-induced cardiomyopathy [16-18], muscle inflammatory foci are mostly composed of T lymphocyte. However, in DMD patients and DMD animal models, lymphoid cells compose a minor inflammatory population in damaged muscles. In fact, it is still poorly understood why myeloid cells such as macrophages are the main inflammatory cell type in dystrophin-deficient muscles. We observed previously that blood T lymphocyte from twelve-week old mdx mice lack membrane CD62L due to P2X7-dependent shedding and attributed the reduced lymphoid migration to muscles to the downregulation of this selectin. However, it is now known that the CD62L is not required for cell transmigration of effector and effector memory T lymphocytes and we decided to revisit and extend the phenotype analysis of the cells targeted for CD62L shedding. We confirmed that mdx mice are enriched in a blood subpopulation of T lymphocyte that lack CD62L; however, this subpopulation is also deficient in CD44 due to P2X7 activity. About fifteen percent of blood CD3+ cells are CD62L- CD44- in mdx mice, a five-fold increase when compared with age-matched C57BL/10 mice. Therefore, we attribute the reduced lymphoid transmigration to damaged muscles to a broader phenotypic alteration, with the lack of mainly CD44.
This molecule plays multiples roles in inflammatory diseases and tumors, as indicated by studies using CD44-null mice and loss-of-function experimental treatments [13]. The molecular engagement to HA on endothelial cells surface results in T lymphocytes rolling, transmigration, and chemotaxis towards inflammatory stimuli. However, it remains to be elucidated why mdx mice respond normally to infections and are not more susceptible to tumors, for example. Regarding the CD69 labeling, our results indicate that more than 10% of the CD62L- CD44- are early activated T lymphocytes (CD127- CD69+). Although we do not know what antigens could have primed these cells, it is interesting to depict a cellular phenotype that is compatible with activated T lymphocyte, but with possible limited capacity to migrate to injured sites, due to the lack of CD44.
We included the analysis of CD127 to help categorizing the CD62L- subpopulation as naϊve, effector or memory T cells. However, and to our surprise, this molecule was also down regulated by P2X7 activity. Therefore, we still do not know if this is an antigen-primed subpopulation and the phenotype as CD62LCD44- CD127- does not fall in any of the known divisions of T lymphocyte and additional experiments are needed to evaluate the origin and function of these cells. We are just starting to understand why T lymphocyte subpopulations downregulate homing/adhesion molecules due to P2X7 activity and migrate less to muscles in DMD. This is particularly important because T lymphocyte can potentially recognize dystrophin epitopes in patients submitted to exon skipping treatment, for example, and be cytotoxic/pathogenic against newly regenerated dystrophin positive myofibers.
Acknowledgments
The authors would like to thank Dr. Gabriel Melo de Oliveira for invaluable support in mice management.
Financial Support:
CNPq grant numbers 407711/2012-0 and 421803/2017-7; Instituto Oswaldo Cruz, Fundação Oswaldo Cruz.
References
- Hoffman EP, Brown RH, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus Cell 51(6): 919-928.
- Blake DJ (2002) Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev 82(2): 291-329.
- Rosenberg AS (2015) Immune-mediated pathology in Duchenne muscular dystrophy. Sci Transl Med 7(299): 299rv4.
- Juban G, Chazaud B (2017) Metabolic regulation of macrophages during tissue repair: insights from skeletal muscle regeneration. FEBS Lett 591(19): 3007-3021.
- Wehling M, Spencer MJ, Tidball JG (2001) A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol 155(1): 123-131.
- Spencer MJ, Spencer MJ, Tidball JG (2001) Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin Immunol 98(2): 235-243.
- Spencer MJ (1997) Myonuclear apoptosis in dystrophic mdx muscle occurs by perforin-mediated cytotoxicity. J Clin Invest 99(11): 2745-2751.
- Cascabulho CM (2016) Chemotaxis and Immunoregulatory Function of Cardiac γδ T Cells in Dystrophin-Deficient Mice. J Immunol 197(9): 3531-3544.
- Cui G (2015) IL-7-Induced Glycerol Transport and TAG Synthesis Promotes Memory CD8+ T Cell Longevity. Cell 161(4): 750-761.
- Chang JT, Wherry EJ, Goldrath AW (2014) Molecular regulation of effector and memory T cell differentiation. Nat Immunol 15(12): 1104-1115.
- Cascabulho CM, Cotta de Almeida V, Henriques-Pons A (2012) Defective T-lymphocyte migration to muscles in dystrophin-deficient mice. Am J Pathol 181(2): 593-604.
- Thorne RF, Legg JW, Isacke CM (2004) The role of the CD44 transmembrane and cytoplasmic domains in co-ordinating adhesive and signaling events. J Cell Sci 117(Pt 3): 373-380.
- Ponta HL, Sherman, Herrlich PA (2003) CD44: from adhesion molecules to signaling regulators. Nat Rev Mol Cell Biol 4(1): 33-45.
- Jordan AR (2015) The Role of CD44 in Disease Pathophysiology and Targeted Treatment. Front Immunol 6: 182.
- Hertweck MK, Erdfelder F, Kreuzer KA (2011) CD44 in hematological neoplasias. Ann Hematol 90(5): 493-508.
- Schmidt J (2018) Current Classification and Management of Inflammatory Myopathies. J euromuscul Dis 5(2): 109-129.
- De Oliveira GM (2007) Fas ligand-dependent inflammatory regulation in acute myocarditis induced by Trypanosoma cruzi infection. American Journal of Pathology 171(1): 79-86.
- Baaten BJ, CR Li, Bradley LM (2010) Multifaceted regulation of T cells by CD44. Commun Integr Biol 3(6): 508-512.
