 Mini Review
Mini ReviewIntroduction
Pathologists Juan Rosai and Ronald Dorfman initially described in 1969 “sinus histiocytosis with massive lymphadenopathy” after they observed symptomatic extensive lymph node enlargement; and subsequently came to be designated “Rosai –Dorfman” disease [1].Rosai-Dorfman disease is a rare disorder characterized by increased production and accumulation of non-Langerhans sinus histiocytes within lymph nodes, most commonly affecting the cervical lymph node chain. Histiocytes may also accumulate in extranodal areas including the skin; central nervous system, kidney, and digestive tract.43% of cases of Rosai Dorfman disease demonstrate extranodal involvement. With skin being the most commonly site [2]. Thawerni et al were the first to report a case of cutaneous Rosai-Dorfman disease in 1978 [3].
Clinical Review
The aetiology of cutaneous Rosai-Dorfman disease is still unknown, immune and viral causes have been hypothesized. The signs and symptoms of this disease depend on which parts of the body are affected. Skin lesions seen in cutaneous Rosai-Dorfman disease are indurated firm papules or nodules measuring between 1-10 cm [1]. Age groups affected include children, adolescents and young adults [4]. The disease presents more commonly in males and in African descent [5]. Cutaneous Rosai-Dorfman disease (without nodal involvement) typically occurs at a later age of onset with a median age of 43.5 years and demonstrates female predominance with a ratio of (2:1), Asian and Caucasian individuals are mostly affected [6]. Laboratory findings are not specific and include: leucocytosis, elevated sedimentation rate and polyclonal gammopathy, elevated serum ferritin and normochromic/normocytic and autoimmune haemolytic anemia [7]. In our experience we recently encountered a patient who is sixty-eight-year-old gentleman of black Caribbean origin with clinical signs and histology suggestive of Cutaneous Rosai-Dorfman syndrome. His past medical history included: insulin dependent diabetic, Hypertension, chronic kidney disease stage 3, recurrent anterior uveitis, sickle cell trait and anaemia. There is no history of fever, night sweats, weight loss, fatigue and ECOG performance status of 0. On examination there is no evidence of any enlarged lymph node clinically, no evidence of organomegally, chest, cardiovascular and neurological examination is grossly normal. Laboratory findings include low HB, Thrombocytosis, high ESR, high CRP, low albumin, normal Iron, raised globulins. He presented with skin hyperpigmentation on his arms and legs for 1-2 years which became more noticeable during the last couple of months. He then presented with asymptomatic lesion on the face, upper limbs and trunk (Figures 1-6). Examination revealed multiple popular nodular lesions on the face and abdomen and arms. Some are typical of molluscum contagiosum, but some are different.
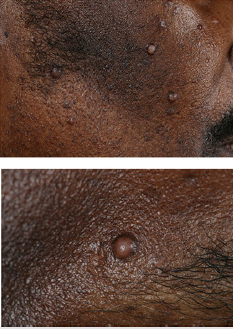
Figure 1: A-Raised nodular hyperpigmented lesions on the face with post-inflammatory hyperpigmentation. B-Close up to one of the raised nodules.

Figure 2: Papular eruption with hyperpigmentation legs.
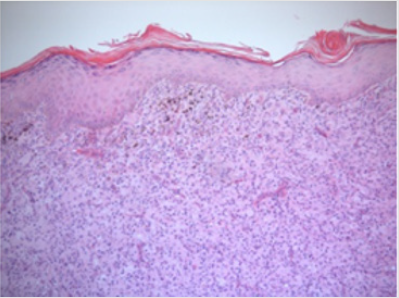
Figure 3: Low power (x4 magnification) H& E skin nodule in which the dermis shows diffuse sheets of histiocyte-like cells.
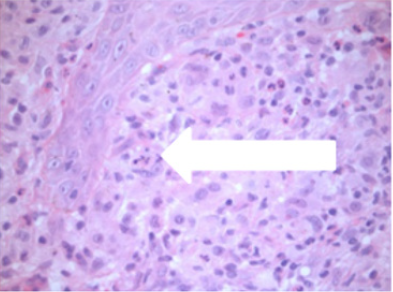
Figure 4: High power (x 40 magnification) demonstrating emperipolesis by histiocytes.
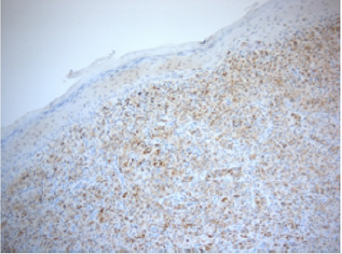
Figure 5: CD68 stain showing scattered positive cells within the dermis.
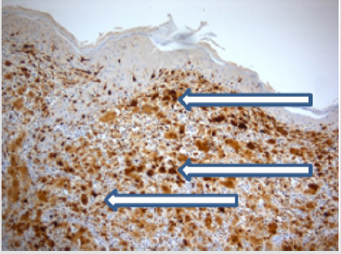
Figure 6: S100 (x 10 magnification) is strongly nuclear positive in the dermal histocytic population
Treatment
Cutaneous Rosai Dorfman syndrome is a self-limiting disease. Treatment options reported in literature include surgical excision is recommended when lesion are localized, cryotherapy was successfully used for cutaneous Rosai-Dorfman syndrome that was refractory to topical and intralesional steroids [8].Dapsone was found to be effective in the treatment of cutaneous Rosai-Dorfman syndrome [9] ,100mg daily of dapsone was used to treat a patient with resistant to isotretinoin and oral, intralesional steroid with complete regression after 3 months of treatment with dapsone and patient remained in remission on his 1 year follow up [10]. Acitretin treatment for 4 months was found to be effective in a 12 year old patient [11,12]. High doses of Thalidomide (300 mg/d) can be effective in cases of extensive disease if patients can tolerate its side effects [13]. Other forms of treatment include isotretinoin, radiotherapy, Intralesional steroids.
Histology Review
The findings in Rosai-Dorfman disease and Cutaneous Rosai- Dorfman Disease are similar. Dermal infiltrates include large histiocytes, lymphocytes, and plasma cells. The histiocyte known as Rosai-Dorfman cell is considered diagnostic of Rosai-Dorfman disease, this cell is having an amorphous cytoplasm, large vesicular nucleus with prominent nucleoli and indistinct border [13]. Presence of lymphocytes and plasma cells engulfed within the cytoplasm of histiocytes is referred to as emperipolesis. This population express S-100 and are considered diagnostic although not pathognomonic of Rosai-Dorfman disease. This histiocytes population also may stain positive for CD68, and negative for CD1a [14,15], helping to exclude other differential diagnoses. In our patients shave excisions from right maxilla, left upper and left lower cheek, on separate occasions showed normal epidermis with a dermal based histiocytic lesion admixed with lymphocytes and plasma cells. This consisted of florid numbers of large epithelioid histiocytes with abundant eosinophilic cytoplasm with round nuclei, wrinkled nuclear membranes and small inconspicuous nucleoli. Emperipolesis of small lymphocytes was identified within occasional large histiocytic cells which helped narrow the differential diagnosis. Interspersed single and clusters of small lymphocytes with scattered plasma cells also seen. Scattered mitoses were noted including a single quadripolar form, but there was no necrosis. Whilst the diffuse histocytic population, and admixed inflammatory cells raised the possibility of cutaneous Rosai-Dorfman, in the initial assessment cells showing emperipolesis were difficult to identify and so other differential diagnoses for an histiocytic infiltrate were also considered such as xanthoma and langerhans cell histiocytosis. Immunohistochemistry demonstrated cells to be positive for S100, and variable CD68 positivity, but this population were negative with melanocytic markers including. Melan A and HMB45; other markers undertaken (CD1a, CD56, Factor 13a) were also negative as were special stains to exclude infective aetiology. S100 assisted in identifying emperipolesis as described by Brenn et al. [6] In conclusion the histological and immunohistochemical findings were all supportive of a pathological diagnosis of cutaneous Rosai- Dorfman disease.
Conclusion
Cutaneous Rosai-Dorfman disease is considered a distinct clinical entity of Rosai-Dorfman syndrome [14]. Extanodal Rosai- Dorfman disease without nodal involvement is extremely rare [6]; skin is considered the commonest extranodal site. Histologically both Rosai-Dorfman disease and Cutaneous Rosai-Dorfman disease have similar findings. However, demographically the disease differs in age, gender and ethnic backgrounds. The median age of presentation of cutaneous Rosai-Dorfman disease is 43.5 years while in Rosai-Dorfman disease is 20.6 years. The Cutaneous Rosai- Dorfman disease shows female predominance in contrast the Rosai- Dorfman disease showed male predominance. Population affected with Cutanoues Rosai-Dorfman disease is mostly white and Asian while in Rosai-Dorfman disease it was mostly black and rare in Asians [16]. The diagnosis is made by histopathological examination looking for the presence of histiocytes that stain positive for S-100 and identification of emperipolesis [17]. Cutaneous Rosai-Dorfman syndrome is a benign condition which is self-limiting but may require treatment or surgical excision [18,19].
References
- Histiocytosis Association (www.histio.org).
- Rosai J, Dorfman RF (1972) Sinus histiocytosis with massive lymphadenopathy: a pseudolymphomatous benign disorder. Analysis of 34 cases. Cancer 30(5): 1174-1188.
- Thawerani H, Sanchez RL, Rosai J, Dorfman RF (1978) The cutaneous manifestations of sinus histiocytosis with massive lymphadenopathy. Archives of Dermatology 114(2): 191-197.
- National Organization for Rare Disorders (NORD).
- Lauwers GY, Perez Atayde A, Dorfman RF, Rosai J (2000) The digestive system manifestations of Rosai-Dorfman disease (sinus histiocytosis with massive lymphadenopathy): review of 11 cases. Human pathology 31(3): 380-385.
- Brenn T, Calonje E, Granter SR, Leonard N, Grayson W, et al. (2002) Cutaneous rosai-dorfman disease is a distinct clinical entity. The American Journal of Dermatopathology 24(5): 385-391.
- Cunha BA, Durie N, Selbs E, Pherez F (2009) Fever of Unknown Origin (FUO) due to Rosai-Dorfman disease with mediastinal adenopathy mimicking lymphoma: diagnostic importance of elevated serum ferritin levels and polyclonal gammopathy. Heart & Lung: The Journal of Critical Care 38(1): 83-88.
- Fumerton R, Ball N, Zhou Y (2011) Refractory cutaneous Rosai-Dorfman disease responsive to cryotherapy. Cutis 87(6): 296-299.
- Chang HS, Son S, Cho KH, Lee JH (2011) Therapeutic challenge of dapsone in the treatment of purely cutaneous Rosai-Dorfman disease. Clinical and experimental dermatology 36(4): 420-422.
- Chan C, Chu C (2006) Dapsone as a potential treatment for cutaneous Rosai-Dorfman disease with neutrophilic predominance. Arch Dermatol 142(4): 428-430.
- Mebazaa A, Trabelsi S, Denguezli M, Sriha B, Belajouza C (2007) Extensive purely cutaneous Rosai-Dorfman disease responsive to acitretin. International journal of dermatology 46(11): 1208-1210.
- Fening K, Bechtel M, Peters S, Zirwas M, Darabi K, et al. (2010) Cutaneous rosai-dorfman disease persisting after surgical excision: report of a case treated with acitretin. The Journal of Clinical and Aesthetic Dermatology 3(9): 34-36
- Lu C, Kuo T, Wong W, Hong H (2004) Clinical and histopathologic spectrum of cutaneous Rosai-Dorfman disease in Taiwan. J Am Acad Dermatol 51(6): 931-939.
- Middel P, Hemmerlein B, Fayyazi A, Kaboth U, Radzun HJ, et al. (1999) Sinus histiocytosis with massive lymphadenopathy: evidence for its relationship to macrophages and for a cytokine-related disorder. Histopathology 35(6): 525-533.
- Juskevicius R, Finley JL (2001) Rosai-Dorfman disease of the parotid gland: cytologic and histopathologic findings with immunohistochemical correlation. Archives of Pathology & Laboratory Medicine 125(10): 1348-1350.
- Wang K, Chen W, Liu H, Huang C, Lee W, et al. (2006) Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. The British journal of dermatology 154(2): 277-286.
- Brenn T, Calonje E, Granter SR, Leonard N, Grayson W, et al. (2002) Cutaneous rosai-dorfman disease is a distinct clinical entity. The American Journal of Dermatopathology 24(5): 385-391.
- Farooq U, Chacon AH, Vincek V, Elgart GW (2013) Purely cutaneous rosai-dorfman disease with immunohistochemistry. Indian journal of dermatology 58(6): 447-450.
- Fang S, Chen A (2015) Facial cutaneous Rosai-Dorfman disease: A case report and literature review. Experimental and Therapeutic Medicine 9(4): 1389-1392.
