 Research Article
Research ArticleAbstract
Gene cloning is a way by which taking a piece of DNA from the organism where it naturally occurs and putting it into a cloning host such as the bacterium Escherichia coli. Bacterial cell that contain foreign DNA can express the genetic information and make the gene products. To be able to perform cloning in E. coli, plasmid pHEN6c vector was used to clone cDNA fragment of a mRNA coding for nanobody. E. coli plasmid was extracted, purified and then digested using restriction enzyme PstI and Eco91I. The purity and concentration of the plasmid were 1.84 and 39.2ng/μl respectively. The pHEN6c vector as well as the nanobody cDNA were digested with PstI and Eco91I restriction enzyme then ligated to form a recombinant plasmid. The constructs were transformed into Escherichia coli strain by heat shock method. The cells were cultured overnight, and transformation efficiency was determined as 7867.22 transformants/μg. In this experiment the target sequence amplified, and bands were obtained following 1% agarose gel electrophoresis of the colony PCR products. To determine the size of the cloned cDNA and the pHEN6c vector plots of the relative morbidity against molecular size (log bp) were used and the size were 573 bp and 3842bp for the cDNA and the vector respectively. Cloning techniques holds many promising future researches in genetic engineering and biotechnology. It is therefore important to learn through practical experiment and research of cloning to exploit the best of nature (living organism) to our advantage.
Keywords: Plasmid; Cloning; Nanobody gene
Introduction
The discovery of the structure and role of DNA and the sorting out of the genetic code has led over the last 50 years to an explosion in our understanding of organisms and how they work. In the forefront of this revolution came gene cloning. The term “gene cloning” covers a wide range of techniques that make it possible to manipulate DNA in a test tube and to return it to living organisms where it functions normally. The importance of this technology is that it allows us to isolate any piece of DNA among the millions of base pairs that make up the genome of an organism. This first step is essential for a whole range of scientific and technological studies, ranging from the study of a gene that is instrumental in causing an inherited disease, to the bioengineering of a strain of yeast that produces a useful pharmaceutical product [1]. Gene cloning involves taking a piece of DNA from the organism where it naturally occurs and putting it into a cloning host such as the bacterium Escherichia coli. It is then possible to study the cloned DNA or produce the protein encoded by the gene. For many applications you may want subsequently to transfer the cloned DNA into another organism, but the initial cloning steps are almost always performed in E. coli [2].
Using restriction endonucleases foreign DNA can be inserted into bacterial plasmids and can be replicated). Bacterial cells that contain foreign DNA can express the genetic information and make the gene products. Thus, by cloning these cells, we can learn about the structure and operation of specific gene and gene products as well as rare protein expression. Plasmid transformation into bacterial competent cells is a key technique in molecular cloning to transform the genetic constitution of other organisms [3].
Plasmids the most commonly used vectors for gene cloning and are small circular DNA molecules found in many types of bacteria having an “origin of replication” which directs the replication of the plasmid and ensures that the cloned cell contains many copies of the plasmid which are distributed between the daughter cells when the cell divides [4]. The exact number of copies varies according to the plasmid. If the cloned gene is part of a DNA molecule with an origin of replication, that is, cloned into a plasmid, it will also be copied when the plasmid is copied. Plasmids commonly used in cloning contain a selectable marker, usually an antibiotic resistance gene. This means that we can tell which bacteria contain the plasmid simply by spreading them onto an agar plate containing the antibiotic where growth is appreciated in organism containing plasmid [5].
Plasmids are considered transferable genetic elements, or “replicons”, capable of autonomous replication within a suitable host. Although it is capable of self-replication, it still uses host cell machinery to perform this function. Plasmids provide a mechanism for horizontal gene transfer within a population of microbes and typically provide a selective advantage under a given environmental state. Plasmids may carry genes that provide resistance to naturally occurring antibiotics in a competitive environmental niche, or alternatively the proteins produced may act as toxins under similar circumstances. Plasmids also can provide bacteria with an ability to fix elemental nitrogen or to degrade recalcitrant organic compounds which provide an advantage under conditions of nutrient deprivation [6].
In this experiment basic principles of genetic engineering were used to solve scientific challenges, research and product development. To achieve our objective plasmids were isolated from E. coli bacterial culture and recombinant plasmid containing specific fragments of nanobody gene constructs were then transformed into E. coli cells, conferring ampicillin resistance to host cells. Other basic techniques for analysis of the results using PCR, and agarose gel electrophoresis were also used.
Methodology
Isolation of Plasmid pHEN6c
Isolation of Plasmid pHEN6c was carried out in 7 successive steps: Cells harvesting, cells re-suspension, cells lysis, Neutralisation, column preparation, loading of cleared lysate, washing of column and DNA elution. We harvested the overnight E. coli culture and put its 1 ml into an Eppendorf tube. We centrifuged it in a microcentrifuge at a high speed of 10600 RPM (12,000×g) in 1 minute. After decanting the supernatant, another 1 ml of the E. coli culture was added to the pellet as centrifuged again at high speed in one minute. The resulting supernatant was poured in a waste beaker whereas the pellet was immediately resuspended in 2μl of the re-suspension solution. The mixture was vortexed to ensure an appropriate and complete re-suspension. To lyse the resuspended E. coli pellet, 2μl of the lysis solution was added. The content was gently inverted around 6 to 8 times (instead of using a vortex, as it can cause the shearing of the genomic DNA and consequently the plasmid DNA contamination). The neutralization immediately followed by adding 350μl of the neutralization solution to precipitate the cell debris. The mixture content was then gently inverted 4 to 6 times. The same content was centrifuged at high speed (12,000×g) in 10 minutes. As our supernatant was not containing many floating particles, the second centrifugation was found unnecessary.
The column was prepared by inserting a GenEute Miniprep Binding column into a microcentrifuge tube. A 500 μl of the Column preparation solution was added to the miniprep column and centrifuged at high speed (12,000×g) in 30 seconds to 1 minute. The flowing-through liquid was discarded. The prepared column served for loading the cleared lysate (resulting from neutralization). The cleared lysate was added to the already prepared column and centrifuged at high speed of 12,000×g in 30 seconds to 1 minute. The washing of the column was then performed by adding 750μl of the dilute wash solution to the column; centrifuged at the speed of 12, 000×g for 30 seconds to 1 minute as well.
The flowing- through liquid was discarded in a waste beaker and the column was again centrifuged at high speed ≥12, 000×g in 1 to 2 minutes to remove any excess of ethanol. We finally eluted the DNA by transferring the column to a fresh collection tube and then add 50μl of the elution solution and centrifuged.
Determination of Plasmid DNA Purity, concentration
The determination of the purity (OD 260/OD 280) and concentration (ƞg/μl) of the isolated plasmid DNA from E.coli was done by the use of spectrophotometer (Nanodrop™); with help of the connected computer software the value for both purity and concentration were calculated and generated.
Restriction Endonuclease Digestion of Nanobody Gene and pHEN6c Plasmid
DNA Digestion (pHEN6c plasmid): The determined DNA plasmid concentration, for our subgroup, was low (279.6 ƞg/μl); therefore, we used an average plasmid concentration of 317.57 ƞg/ μl resulting from a combination of the four highest DNA plasmid concentrations (335.6, 353.6, 299.1, and 282 ƞg/μl) obtained by other subgroups. To 11.75μl of distilled water, a volume of 31.25μl (of the DNA plasmid with 317.57 ƞg/μl) was added, followed by the addition of 5μl of 10x Buffer-o (Fermantas) and 1μl of restriction enzyme Eco91I (10units/μl) and restriction enzyme PstI (10units/ μl).The whole mixture (50μl) was then incubated overnight at 37°C.
Digestion and Purification of PCR Amplified cDNA: The cDNA (Nanobody gene PCR fragments) already prepared and purified were subjected to digestion by specific restriction enzymes. To 29.41μl of distilled water, a volume of 13.59μl (of PCR fragments) was added, followed by the addition of 5μl of 10x Buffer-o (Fermantas) and 1μl of restriction enzyme Eco91I (10units/μl) and restriction enzyme PstI (10units/μl).The whole mixture (50μl) was then incubated overnight at 37°C. The GenElute TM PCR Clean-UP Kit came in play for cleaning up (purifying) the PCR amplilified cDNA. We started with preparing the column (to maximize the binding of the DNA to the membrane). A volume of 0.5 ml of the column preparation solution was added to the a GenElute Miniprep Binding Column already inserted into a collection tube.
The collection tube was then centrifuged at 12, 000 rcf for 30 seconds to one minute. The discard was discarded. A volume of 500μl of binding solution was added and mixed to 50μl of the PCR reaction. The mixture solution was transferred into the binding column. The column (into the collection tube) was centrifuged at 12, 000-16,000 rcf for one minute. The eluting liquid was discarded. The diluted washing solution, 0.5 ml, was added to the column and centrifuged at 12,000-16,000 rcf for one minute. The elute was discarded. To remove any ethanol in excess, the collection tube (with column into) was then centrifuged at 12,000-16,000 rcf for 2 minutes. The collection tube with the elute was discarded; whereas the column was subsequently transferred in a fresh 2 ml collection tube. The elution was performed by the addition of 50μl of the elution solution to the column. To elute the DNA, the column was centrifuged at 12, 000-16, 000 rcf for one minute. The resulting elute was containing the purified PCR product; and it was immediately used (or stored at -20°C).
Ligation
To 13.9 μl of distilled water, 2.28 μl of PCR fragments , 0.82 of plasmid vector , 2μl of ligation buffer (0.2 M Tris pH=7.6, 50 mM MgCl2, 50 mM DTT, 500 ug/ml BSA, 5mM ATP) and 1μl of T4 DNA were added up in one tube to make a total volume of 20μl. The solution was at room temperature for 2 hours.
Transformation
Generation of Cacl2 Competent Cells: To prepare ‘heat shock’ competent cells, 5 ml of LB (without antibiotics) was inoculated with added in one with a single colony from a fresh plate, in one sterile 50 ml tube. The inoculated medium was then incubated at 37°C overnight (with shaking vigorously at 230 rpm). The following morning, 20 ml LB was inoculated with 0.2 ml of the overnight culture. The tube was placed in incubator for 90 to 180 minutes for growing the inoculated bacteria to their early log phase. Later it was transferred to ice for 10minutes. The cells were then collected by centrifugation at 3000 rpm in eppendorf tube, supernatant was discarded and then the pellet was resuspended with 10 ml sterile ice cold 0.1 M MgCl2. The pellet was again re-suspended with a different 10ml sterile ice cold 0.1M CaCl2, followed by its incubation in ice for 1 hour and then centrifuged at 7 min 3000 rpm in 4°C eppendorf centrifuge. The supernatant was poured and sterile ice cold 0.1 M CaCl2 was added plus 0.3 ml sterile ice cold 100 % -glycerol. The mixture was then subject to incubation for 30 min on ice.
Heat Shock Transformation: Three 1.5 ml eppendorf tubes were prepared, each containing 100μl cell suspension, and labeled as T1, T2 and T3 respectively. Among the three tubes, T1, T2 and T2 were prepared as positive control, tube, test tube and negative control tube respectively. Aliquot 0.05μg of purified intact plasmid DNA was added to T1, 10μl of unpurified ligation to T2 and T3 was left with its initial 100μl of Cacl2. The three tubes were incubated on ice for 30 minutes. After their incubation, the three tubes were subsequently transferred to warm bath at 42°C for exactly 90 seconds and then put back on ice for 2 minutes. After incubation, the cells were then cultured on LB agar containing ampicillin by spreading (with a sterile lazy spreader) 100μl of the transformation preparation from each tube on its LB-AMP agar plates (labeled with T1, T2 and T3). The cultured plates were incubated at 37°C upside down overnight. On the next morning (after 16 to 20 hours), the number of colonies was determined on each plate.
Polymerase Chain Reaction (PCR)
The PCR master mix was first prepared. In each PCR tube of the five, 50μl of master mix (5μl of 10x PCR buffer, 1μl of dNTP, 1μl FP, 1μl RP, 0.25μl Taq DNA polymerase, 41.75μl dH2O) was added. Using sterile pipette tips, one colony was picked from the positive test (T1) and mixed in one PCR tube; another one colony was picked from the test tube as there was no growth of any colony on out test plate T2 and was dipped in a separate PCR tube as well. The third tube to which there was no colony added in, served as a negative control. The tubes were all closed and then transferred to a thermo cycler that cycled between melting (at 94°C for 30seconds), annealing (at 57°C for 30seconds) and polymerization temperatures (72°C for 45seconds). The gene fragments encoding nanobody (nanobody cDNA insert), if present in the sample, were amplified with the specific primers: forward primer (FP 5’-TTCCCAGTCACGAC-3’) and reverse primer (RP 5’- CACACAGGAAACAGCTATGAC-3’) Arbabi Ghahroudi, 1997.
Analysis of PCR Product by Agarose gel Electrophoresis
The 1% (w/v) molten agarose gel was first poured in 1x TBE in a tray. We then added 30 μl of ethidium bromide at 10 mg/ml to molten gel and subsequently mixed by stirring with the sterile pipette tip. A comb was then applied on the gel holder and removed all possible air bubbles using a cleaned pipette tip. We let the gel to solidify for 20 minutes. The solid gel with its tray was put in an electrophoresis buffer tank; and then 1xTBE electrophoresis buffer was added to completely cover the gel. To each 5μl of the sample PCR product, 2μl loading buffer were added; whereas as for the negative control, only 1μl of the PCR product was mixed with 1μl loading buffer. Both negative and positive control were mixed with 4μl loading dye and then loaded on the gel. Positive mixture was loaded from the second slot onwards; and the negative control mixture was loaded in the last slot. We loaded 10 μl smart ladder on the first slot.
We put the lid on the buffer tank and then connected the positive and negatives electrodes with a voltage of 125V for 1 hour. The gel was then observed under UV light for the separation of bands and compared with the marker (Hyladder™ 10kb) loaded in lane1.The size of the DNA vector was calculated by employing linear relationship of logarithm of base pair and relative mobility along the electric field.
Molecular Weight Determination of DNA after Resolution by Gel Electrophoresis
The molecular weight of nanobody cDNA and plasmid (vector) was determined by 1% agarose gel electrophoresis.
Results
Plasmid DNA Concentration and Purity
After isolation and purification of pHEN6c plasmid DNA, its concentration and purity were determined by Nano Drop™. Samples were then analyzed using a blank reference consisting of water which was used as elution buffer. As for the pure DNA A260 /A280 value should be between 1.7-1.9, thus the plasmid DNA was isolated in its pure form. Our pooled DNA concentration was 317ng/μL which is and pooled purity of 1.84 and the yield of our DNA in 50 μL was 371 μg/ μL multiplied by 50 μL which is 18.5 μg.
Digested cDNA Concentration
Concentration of digested cDNA was 110.3ng/ μL which was 0.1103 and the yield of our DNA in 50 μL was found to be 50 μL multiplied by 0.1103 μg/ μL which is 5.5 μg.
Determination of Molecular weight of cDNA
For molecular weight determination of the cDNA, it was run on 1 % agarose gel and following bands were obtained and the Molecular weight of the cDNA is to be 573 bp (Figure 1).
Determination of molecular weight of Plasmid DNA (vector)
For molecular weight determination of the cDNA, it was run on 1 % agarose gel and following bands were obtained and the Molecular weight of the cDNA is to be 3842 bp (Figure2).

Figure 1: Bands obtained for the cDNA on 1% agarose gel electrophoresis.
KEY: L-ladder, S1-sample 1, s2-sample2, s3-sample 3, S4- sample 4, S5-sample 5, S6-sample 6
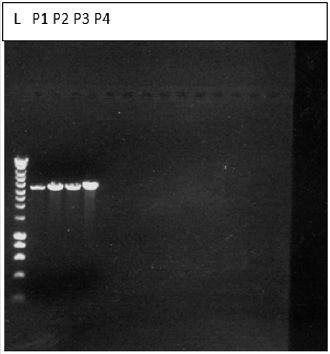
Figure 2: Bands obtained for the Vector DNA on 1% agarose gel electrophoresis.
KEY: L-ladder, P-plasmid
Determination of Efficiency of Transformation
Transformation efficiency is a measure of the amount of cells within the bacterial culture that are able to take up DNA molecules. Calculation was started with the amount of intact plasmid DNA plated: The number of colonies was found to be 349 cfu (Figure 3) in 50 ng (0.05 μg) of DNA in 1μl was added to 100 μl cells. The concentration of DNA in the solution = (0.05 μg /101μl) ≈ 0.00049 μg/μl. Then, 100μl of culture was added to each of the plates, therefore amount of intact plasmid DNA plated on each plate was calculated as: 0.05/1101= 4.54x10-5 μg/μl x 100 μl = 4.54x10-3 μg.
Amount of DNA plated (μg/ml): 4.54x10-3 μg /100μl=4.54x10-3 μg /0.1ml = 4.54x10-2 μg /ml. Using the formula (Transformation efficiency= Number of colonies on LB-AMP plate divided to amount of DNA plated), it was calculated as 349cfu/4.54x10-2 μg /ml = 7687.22cfu/1μg/ml.

Figure 3: Colony growth on T2.
Determination of Percentage of Transformants Having the Insert
After doing colony PCR, the amplified samples from one randomly selected colony along with common positive and negative control were run on 1% agarose gel and no or faint and were obtained under UV exposure.
Optical Density (OD) Measurement of Cell
Optical Density (OD) was measured for Cells grown at a log phase by spectrophotometer at OD 600 and the result is 0.213 where the standard lies between 0.2 to 0.4.
Growth at LB+Ampicilin
Growth of colonies was seen in positive and test plate, but no growth of single colony was found at the third sample which is negative control (Figures 4 & 5).
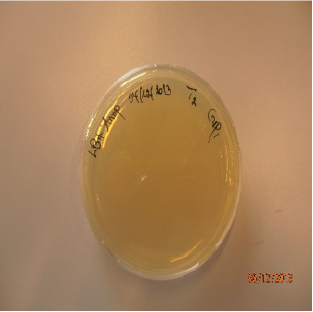
Figure 4: No colony growth on T2.
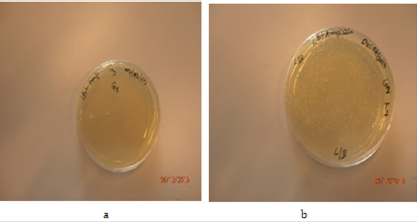
Figure 5: Colonies growth on both T1 (Positive control) and absence of colonies growth on T3.
Discussion
The concentration obtained from Nanodrop spectrophotometer (18.5 μg/50 μl) from 2ml culture of E.coli containing pHEN6c indicates that plasmid DNA isolation via alkaline denaturation is effective method. Our result agrees with the findings of Sabine (2003)[7],using the procedure alkaline denaturation and yield of 2-5μg DNA from a 1.5-mL culture of E.coli containing a pBR322- derived plasmid, and three- to five-fold higher yields can be expected from pUC-derived plasmids. Transformation efficiency represents the total number of bacterial cells that contains the inserted DNA. In our experiment the transformation efficiency was high at 7687.22cfu/1μg/ml transformants/ μg compared to Baraka [8] who got 1.107 x 102 transformants/ μg .This efficiency implies that our plasmids have been successfully uptaken by the host bacteria which enabled growth on the medium supplemented with ampicillin. The β-lactamase protein is produced and secreted by bacteria that carries the inserted plasmid construct inactivating the ampicillin present in the LB agar, allowing bacterial growth [9]. Only transformed bacteria that contain the plasmid and express β-lactamase grew on plates which contain ampicillin [10].
Untransformed cells like the plate number two of our sample could not grow on the ampicillin selection plates hence enabling selection of only positive transformants. Some factors that might have influenced the transformation efficiency are the addition of calcium chloride, which induced the bacterial cell to take up the DNA by increasing its membrane permeability. The amount of naked DNA placed into the bacterium’s environment might also play a role, the more DNA, the more efficient the transformation would be [11]. The PCR product were resolved on 1% agarose gel and faint band could be detected both in the samples and the positive control as well. Amplification failure might be due to PCR primer failure or poor enzyme activity of the polymerase, buffer problem. This is also supported by Roux [12] who states primers sequence could be synthesized or diluted in correctly, buffer and inhibitors were incriminated to affect amplification, to resolve this problem a new design for an optimal primer pair is required to attain required amplification. Colony PCR failure can also be caused by too much cells in the PCR reaction, to avoid this colonies can be diluted in sterile distilled water before running PCR [13].
Conclusion
Since nanobody have good expression in microbial systems and the beneficial biochemical properties (good solubility, good stability in harsh conditions, high affinity and specificity for the antigen, small size and strict monomeric behavior) they are an ideal tool for research purposes or diagnostic or therapeutic applications. Even though the colony PCR in our experiment was not successful (due to buffer and enzyme defect) the Cloning procedure were help full for the insertion of Nanobody gene cDNA into the bacterial genome, E.Coli WK6 strain. Cloning techniques holds many promising future researches in genetic engineering and biotechnology. It is therefore important to learn through practical experiment and research of cloning to exploit the best of nature (living organism) to our advantage.
Declaration
This work has never been submitted any were and it is original in its type and I give full permission for the iiste.org academic journal to publish it.
References
- Freeman, Willard M, Daniel J Robertson, Kent E Vrana (2000) Fundamentals of DNA hybridization arrays for gene expression analysis. Biotechniques 29(5): 1042-1055.
- Lodge J, Lund PA, Minchin S (2007) Gene cloning: principles and applications. Taylor and Francis Group.
- Carson S, Robertson D (2005) Manipulation and expression of recombinant DNA. San Diego: Elsevier academic press.
- Alberts B, Johnson A, Lewis J (2002) Molecular Biology of the Cell, (4th Edition) Garland Science, New York, USA.
- Lodish H, Berk A, Zipursky SL (2000) Molecular Cell Biology, (4th). W Freeman, New York, USA.
- Lipps G (2008) Plasmids: Current Research and Future Trends. Caister Academic Press.
- Sabine E, Dirk S (2003) E coli Plasmid Vectors, Isolation of Plasmids from coli by Alkaline Lysis: Methods in Molecular Biology 235: 75-78.
- Hanahan D (1985) DNA Cloning Techniques: A Practical Approach. In: DM Glover (ed,). (IRL Press, Oxford) 1: 109.
- Campbell, Neil A (2005) Biology. In: Neil A Campbell, Jane B Reece (Eds.), 7th (edn.). CA: Pearson Education. Inc, San Francisco, USA, pp. 384-388.
- Maeda S, Sawamura A, Matsuda A (2004) Transformation of colonial Escherichia coli on solid media. FEMS Microbiology Letters 236(1): 61-64.
- Kenneth H Roux (2001) Optimization and Troubleshooting in PCR Genome. Res 1995 4: S185-S194.
- Lee AB, TA Cooper (1995) Improved direct PCR screen for bacterial Colonies. Bio Techniques 18: 225-226.
- Barakat (2011) practical report of IPMB.
- Primrose SB, Twyman RM, Old RW (2006) Principles of Gene Manipulation. Blackwell Science, Inc: Malden.
- Steven Odongo (2013) Students Training Manual for IPMB.
- Alberts B, Johnson A, Lewis J (2002) Molecular Biology of the Cell. 4th Edition, Garland Science, New York, USA.
