 Research Article
Research ArticleAbstract
Paracetamol or acetaminophen is a popular pain and fever releasing and wellknown over-the-counter drug. It metabolized by different hepatic pathways. High and long-term doses can cause hepatotoxicity and nephrotoxicity. It suppresses the prostaglandin synthesis by blocking the cyclooxygenase. Attempt has been taken to investigate the physicochemical, binding affinity, and pharmacokinetic properties of some major PCT metabolites. Density functional theory (DFT) along with B3LYP/6-31G basis set has been employed to optimize the structures. Molecular docking simulation has been performed against prostaglandin synthase protein (5F19) to compare the binding capability and ADMET prediction has been utilized to predict the absorption, metabolism and toxicity.
Keywords: Paracetamol; Metabolites; Physicochemical Properties; Molecular Docking; ADMET
Abbreviations: PCT: Paracetamol; NAPQI: N-acetyl-p-Benzoquinone Imine; MEP: Molecular Electrostatic Potential; BBB: Blood Brain Barrier; DFT: Density functional theory
Introduction
Paracetamol (PCT) is a mostly used analgesic and antipyretic drug [1]. Overdoses can cause of liver and kidney damage [2]. The primary metabolism of PCT occurred in the liver and some of the metabolites show toxic interaction with the amino acid residue of receptor protein [3]. Paracetamol sulphate (PCT-S) is a primary metabolite, which is formed by sulphate conjugation of PCT and mainly found in children. Similarly, paracetamol glucuronide (PCT-G) is another primary metabolite, which is also formed by glucuronide conjugation of PCT and mainly found in adult. N-acetylp- benzoquinone imine (NAPQI) is one of the toxic metabolites, produced by cytochrome P450 (CYP450) mediated N-hydroxylation of PCT [4–7]. Till now, the metabolite formation and their action are not completely known. Previously, some of the metabolites were detected and reported by many researchers [3,8]. Attempt has taken to optimize the reported metabolites to make more understandable about the metabolite’s formation, their inherent stability, chemical reactivity, binding affinity, and toxic parameters. The enthalpy, free energy, dipole moment, molecular orbital, chemical hardness, softness, electrostatic potentiality has been reported to compare the inherent stability, chemical reactivity. In addition, molecular docking has been performed against prostaglandin synthase protein (5F19) to compare their binding affinity, and mode(s). ADMET prediction has been performed to compare the absorption, metabolism, and safety level for oral administration of all optimized structures (Figure 1).
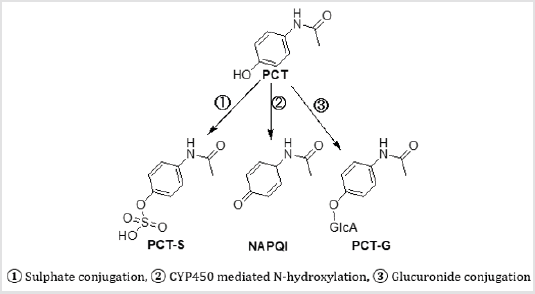
Figure 1: Some major metabolites of paracetamol.
Computational Details
Initial geometry of Paracetamol and its metabolites were collected from online chemical structure database named ChemSpider [9] and geometry optimization done by Gaussin03 software [10]. Density functional theory (DFT) along with B3LYP/6-31G basis set has been employed to optimize the geometry. Molecular orbital features HOMO (Highest occupied molecular orbital), LUMO (Lowest unoccupied molecular orbital) were calculated at the same level of theory. For every metabolite, HOMO-LUMO energy gap, chemical hardness, and softness were calculated from the energies of εHOMO, εLUMO by using the following equation;
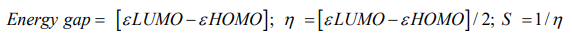
The 3D structure of prostaglandin synthase protein (PDB ID: 5F19) was obtained in pdb format from online protein data bank (PDB) database [11]. All hetero atoms, water molecules, and inactive chain were erased by using PyMol (version 1.3) software packages [12]. The energy minimization of protein was done by Swiss-Pdb viewer software (version 4.1.0) [13]. Finally, the docking simulation was performed by PyRx software (version 0.8 ) [14] considering the protein as macromolecule and drug as ligand with the center grid box size 20.8612, 37.5501 and 59.3402 Å along x, y and z directions respectively. AdmetSAR online database was utilized to predict the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of all metabolites [15].
Result and Discussion
Thermodynamic Analysis
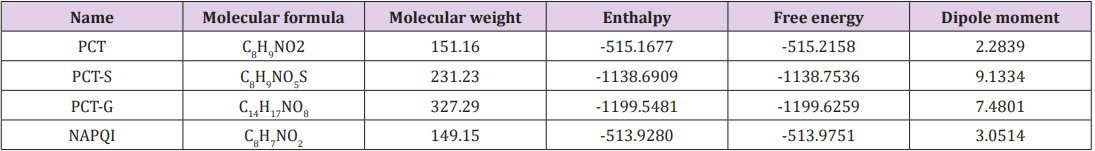
Table 1: Molecular formula, molecular weight, enthalpy, electronic energy, free energy (in Hartree), and dipole moment (Debye) of Paracetamol (PCT) and its modified derivatives.
Thermodynamic properties such as enthalpy of formation and Gibb’s free energy of a reaction help us to predict the spontaneity of the reaction and stability of PCT metabolites [16]. Free energy is a crucial criterion to represent the interaction of binding partners where both the sign and magnitude are important to express the prospect of biomolecular incidents occurring [17]. Negative free energy values favorable for spontaneous binding. In adduct form all PCT metabolites have more negative free energy and enthalpy values compared to PCT. Appreciable changes are observed for PCT-G and PCT-S, hence suggesting sulphate conjugation, CYP450 mediated N-hydroxylation, Glucuronide conjugation are energetically and configurationally more preferable. The highest free energy change is observed for PCT-G. Here, the addition of glucuronide at the replacement of H in –OH of PCT changes the free energy of the structure from -515.2158 Hartree to -1199.6259 Hartree and replacement of H of PCT with sulphate conjugation changes the free energy to -1138.7536 Hartee. By CYP450 mediated N-hydroxylation of PCT, free energy reduces to -513.9280 Hartee. Dipole moment of a drug structure is a significant indicator of drug-receptor interaction [18]. Polar nature of a compound promotes in forming hydrogen bond interactions. All modified PCT metabolites shows different dipole moment compares to PCT. PCT-S and PCT-G shows increased dipole moment (9.1334 Debye and 7.4801 Debye) compared to PCT (2.2839 Debye) and NAPQI (3.0514 Debye) which shows that they have increasing binding affinity against 5F19 (Table 1).
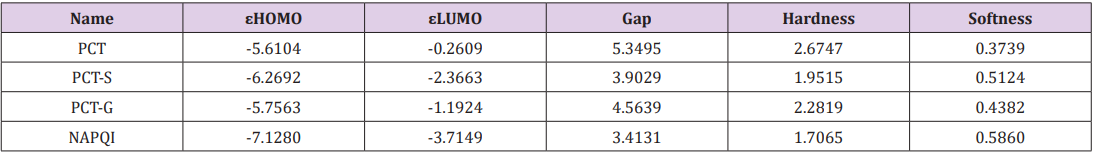
Table 2: Energy (eV) of HOMO, LUMO, energy gap, hardness, softness and chemical potential of (PCT) and its derivatives.
Molecular Orbital Analysis
The electronic absorption relates to the transition to the first excited from the ground state and mainly described by one electron excitation from HOMO to LUMO. The HOMO-LUMO energies, hardness, softness, chemical potential of all drugs is presented in Table 2. Determination of frontier orbital energy gap can be helpful to dictate the kinetic stability and chemical reactivity of a compound kinetic stability increase with the increase of HOMOLUMO gap. As a result, removal of electrons from ground state HOMO to excited state LUMO requires more energy. No increased orbital gap is observed in PCT-G, PCT-S and NAPQI compared to PCT. Sulphate conjugation, Glucuronide conjugation and CYP450 mediated N-hydroxylation show little effect on orbital energy gap values of both low and high molecular weight structures as seen in PCT metabolites respectively. PCT-S (0.5124), PCT-G (0.4382) and NAPQI (0.5860) show higher softness, which may contribute to their chemical activity and polarizability than PCT (0.3739) (Figure 2).
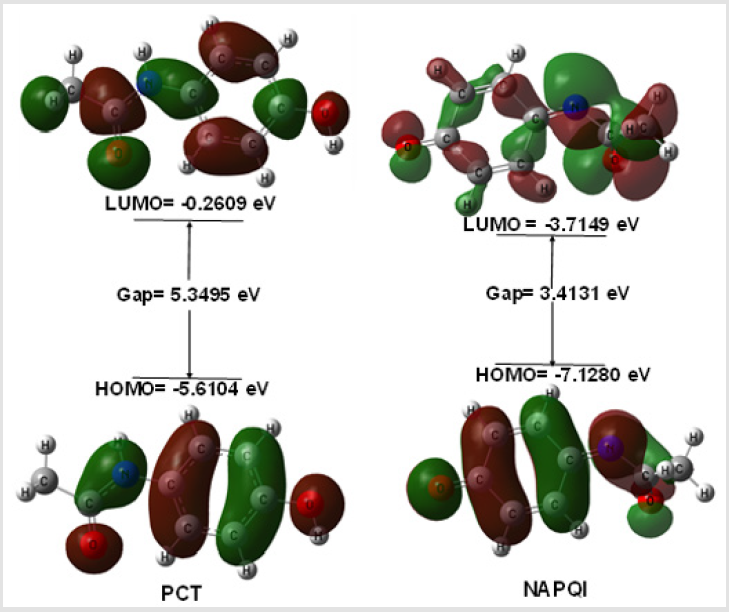
Figure 2: HOMO-LUMO pictures and related energy gap of PCT and NAPQI.
Molecular Electrostatic Potential Analysis
Molecular electrostatic potential (MEP) was calculated at B3LYP/6-31G (d) level of theory to figure out the reactive sites for electrophilic and nucleophilic attack of all optimized structures. The different values of the electrostatic potential are represented by different colors at the surface; red represents regions of most electronegative electrostatic potential, blue represents regions of the most positive electrostatic potential and green represents region of zero potential [19]. MEP displays molecular size, shape as well as positive, negative and neutral electrostatic potential regions simultaneously in terms of color grading. From the MEP map it is clearly seen region having the negative potential are over electronegative atom (Oxygen atom) of C=O functional group in PCT, in sulphate conjugation part of PCT-S [20]. Mainly in all metabolites, the O atom is having the most negative region. The region having positive potential are over hydrogen atoms. Here, the maximum negative potentiality is found in PCT-G (-0.260 a.u) for oxygen atom and highest positive potentiality of PCT-S is (+0.490 a.u) (deepest blue) of hydrogen atoms in PCT-S which suggests the maximum possibility for the nucleophilic and electrophilic attack to that region (Figure 3).
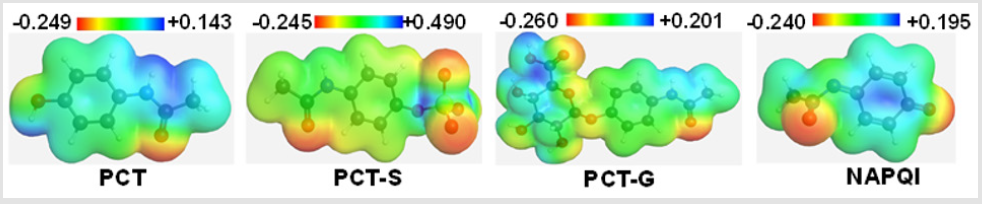
Figure 3: Molecular electrostatic potential map of PCT and its major metabolites.
Molecular Docking and ADMET Analysis
Binding affinities are summarized in Table 3. Greater negative values of binding affinity indicate stronger binding between the receptor protein and the PCT metabolites. The binding affinity of PCT is -6.5 kcal/mol. After sulphate conjugation, the binding affinity of the metabolite PCT-S has increased to -6.9 kcal/mol and glucuronide conjugation in metabolite PCT-G has also increased its binding affinity to -8.6 kcal/mol which is remarkably high with compare to PCT. The glucuronide and sulphate conjugation of PCT-G and PCT-S have not only increased the physicochemical properties but also binding affinity and specialty. In our study, all the compounds show several significant hydrogen bonds. Strong hydrogen bonding is the most symbolic contributing factor in increasing binding affinity of the metabolites of PCT with the receptor (Figure 4) [8]. All the PCT and its metabolites show multiple nonbonding interactions after the docking with 5F19. Non-covalent interaction such as hydrogen bond and hydrophobic interaction are involved in the binding of metabolites. Superimposed view of docked conformation of PCT-G and PCT-S has been shown in Figure 4 where it clearly indicated that due to higher binding affinities, PCT-G and PCT-S has strong affinity to the docking pocket of receptor 5F19 with their present hydrogen bonds present in –NH,–OH and –CH3 group. Finally, we summarized and discussed the reported drug–protein interactions between paracetamol and 5F19 especially with the PCT metabolites. Overall, we are able to provide new insights into the structural and functional roles of paracetamol and its metabolites that can inform the potential prevention and treatment of paracetamol overdose (Figure 4).
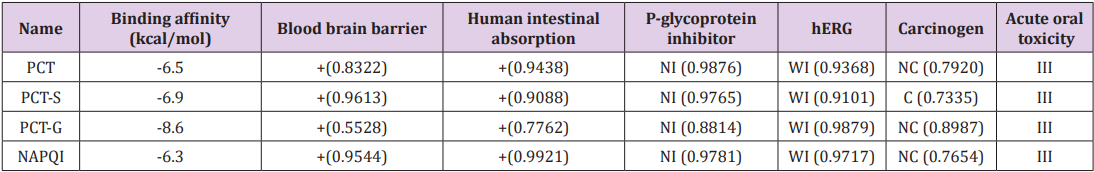
Table 3: Selected pharmacokinetic parameters of PCT and its derivatives.
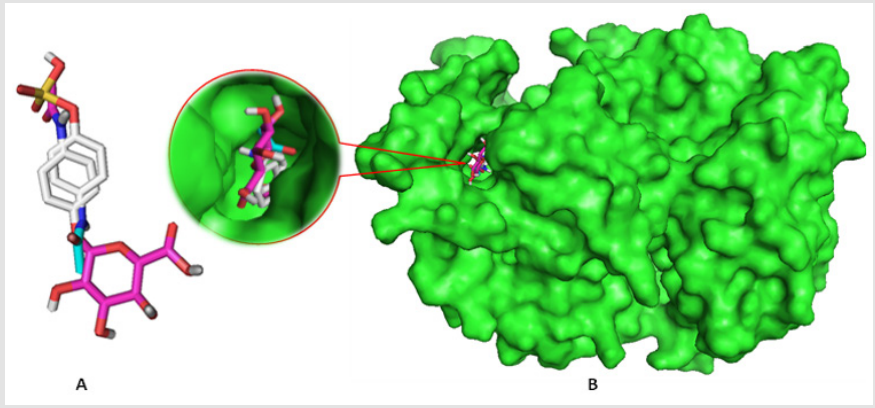
Figure 4: a) Superimposed view of PCT-G and PCT-S
b) Docked conformation of them at the inhibition bounding site of 5F19.
Pharmacokinetic Properties
Admet SAR online database was utilized for the prediction of absorption, inhibition, metabolism and toxicity of paracetamol metabolites [21]. Presumed pharmacokinetic values are listed below in Table 3. The calculation predicts all PCT metabolites are non-carcinogenic except PCT-S, which is the primary metabolite of PCT by sulphate conjugation by sulfotransferase enzyme, and in particular, its production is not significantly higher after oral administration. All the PCT metabolites possess category III oral toxicity, so the metabolites can be predicted to be relatively harmless in case of oral administration [22]. The metabolites show positive response for blood brain barrier (BBB) issue and predict that metabolites will go through BBB easily. NAPQI is predicted to be non-carcinogenic and has positive value in BBB and intestinal absorption but is has higher toxicity level than other metabolites but due to its small amount of production, it is detoxified by conjugation of glutathione and it’s depletion also reduces the capacity to detoxify N-acetyl-pbenzoquinone imine (NAPQI), the toxic metabolite of acetaminophen (PCT) [23]. In the evaluation of acute oral toxicity of PCT metabolites, it is observed that it possesses category III oral toxicity. However, metabolites of PCT have shown weak inhibition of Human Ether-a-go-go-Related Gene (hERG). Inhibition of hERG can lead to long QT syndrome, so further analysis on this aspect is necessary. All the metabolites are non-inhibitor of P-glycoprotein, which is also known as multidrug resistance protein-1.
Conclusion
All the metabolites of PCT are thermally and configurationally more stable than PCT (paracetamol). Most of them have higher softness values, which means higher chemical reactivity with lower HOMO-LUMO gap. From the electrostatic potential (ESP) studies, it is observed that almost all the metabolites and PCT has electrophilic and nucleophilic site for binding in their O and H atom. From docking arrangement, PCT-S, PCT-G, and NAPQI show higher binding affinity than PCT, where most significant interactions are observed for PCT-G with receptor 5F19 and it helped to find amino acid residues that play major role in drugreceptor interaction. Sulphate and glucuronide conjugation, CYP450 mediated N-hydroxylation of PCT increase its binding affinity with 5F19. Relatively small radial volume of glcA group in PCT-G allows it to bury deeper into hydrophobic pocket of protein with highest binding affinity (-8.6 kcal/mol) and form hydrogen bonds. All the pharmacokinetic properties show positive values for the PCT metabolites. Pharmacokinetic calculation predicts all drugs are non-carcinogenic except PCT-S. The metabolites show positive response for BBB and human intestinal absorption and show type III oral toxicity which means they are safe for oral consumption. The metabolites are weak inhibitor for hERG.
Conflict of interest
Authors declare no conflict of interest.
References
- Bertolini A, Ferrari A, Ottani A (2006) Paracetamol: new vistas of an old drug. CNS Drug Rev 12(3-4): 250-275.
- Thomas SHL (1993) Paracetamol (acetaminophen) poisoning. Pharmacol Ther 60: 91-120.
- El-Shahawy A (2014) DFT cancer energy barrier and spectral studies of aspirin, paracetamol and some analogues. Comput Chem 2(1): 6-17.
- Prescott LF (1980) Kinetics and metabolism of paracetamol and phenacetin. Br J Clin Pharmacol 10: 291S-298S.
- Wang Y, Lin W, Wu N (2018) An insight into paracetamol and its metabolites using molecular docking and molecular dynamics simulation. J Mol Model 24(9): 243.
- Jóźwiak Bebenista M, Nowak JZ (2014) Paracetamol: mechanism of action, applications and safety concern. Acta Pol Pharm 71(1): 11-23.
- Prescott LF (2000) Paracetamol: past, present, and future. Am J Ther 7(2): 143-147.
- Uzzaman M, Shawon J, Siddique ZA (2019) Molecular docking, dynamics simulation and ADMET prediction of Acetaminophen and its modified derivatives based on quantum calculations. SN Appl Sci 1: 1437.
- Pence HE, Williams A (2010) ChemSpider: An Online Chemical Information Resource. J Chem Educ 87: 1123-1124.
- Frisch MJ, Nielsen AB (2003) Gaussian 03 Programmer’s Reference. Gaussian.
- Berman H, Henrick K, Nakamura H (2003) Announcing the worldwide protein data bank. Nat Struct Mol Biol 10(12): 980.
- DELANO WL (2002) The PyMOL Molecular Graphics System. De-Lano Scientific, San Carlos, CA, USA.
- Guex N, Peitsch MC (1997) SWISS‐MODEL and the Swiss‐Pdb Viewer: an environment for comparative protein modeling. Electrophoresis 18(15): 2714-2723.
- Dallakyan S, Olson AJ (2015) Small-Molecule Library Screening by Docking with PyRx. In Hempel JE, Williams CH, Hong CC (eds.), Chemical Biology: Methods and Protocols. Springer New York, New York, NY, Pp: 1263: 243-250.
- Van De Waterbeemd H, Gifford E (2003) ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov 2(3):192.
- Loew GH, Goldblum A (1985) Metabolic activation and toxicity of acetaminophen and related analogs. A theoretical study. Mol Pharmacol 27(3): 375-386.
- Emslie KR, Calder IC, Hart SJ, Tange JD (1981) Induction of paracetamol metabolism in the isolated perfused kidney. Xenobiotica 11(9): 579-587.
- Gleeson MP, Gleeson D (2009) QM/MM Calculations in Drug Discovery: A Useful Method for Studying Binding Phenomena? J Chem Inf Model 49(3): 670-677.
- Blaney F, Finn P, Phippen R, Wyatt M (1993) Molecular surface comparison: Application to drug design. J Mol Graph 11(2): 98-105.
- Suvitha A, Periandy S, Boomadevi S, Govindarajan M (2014) Vibrational frequency analysis, FT-IR, FT-Raman, ab initio, HF and DFT studies, NBO, HOMO-LUMO and electronic structure calculations on pycolinaldehyde oxime. Spectrochim Acta Part A Mol Biomol Spectrosc 117: 216-224.
- Lin JH, Lu AYH (1997) Role of Pharmacokinetics and Metabolism in Drug Discovery and Development. Pharmacol Rev 49(4): 403-449.
- Walum E (1998) Acute Oral Toxicity. Environ Health Perspect 106: 497-503.
- Vermeulen NPE, Bessems JGM, Straat R Van De (1992) Molecular Aspects of Paracetamol-Induced Hepatotoxicity and its Mechanism-Based Prevention. Drug Metab Rev 24(3): 367-407.
