 Case Report
Case ReportAbstract
Pantothenate kinase-associated neurodegeneration (PKAN), formerly called Hallervorden-Spatz syndrome, is a rare, inherited neurological movement disorder characterized by the progressive degeneration of specific regions in the central nervous system (neurodegenerative disorder). PKAN is the most common type of neurodegeneration with brain iron accumulation (NBIA), a group of clinical disorders marked by progressive abnormal involuntary movements, alterations in muscle tone, and postural disturbances (extrapyramidal). These disorders show radiographic evidence of iron accumulation in the brain. PKAN is typically diagnosed by molecular genetic testing, most often after a characteristic finding on magnetic resonance imaging (MRI), called the “eye-of-the-tiger” sign, is detected.
Abbreviations: PKAN: Pantothenate Kinase-Associated Neurodegeneration; NBIA: Neurodegeneration with Brain Iron Accumulation; MRI: Magnetic Resonance Imaging
Overview of Hallervorden-Spatz Syndrome
Hallervorden-Spatz syndrome, also known as pantothenatedependent neurodegeneration, is a genetic disorder in the nervous system that is characterized by progressive motor impairment and usually begins in childhood. Movement disorders include involuntary muscle spasms, muscle stiffness, and walking problems that worsen over time [1].
Clinical Signs and Symptoms of Hallervorden-Spatz Syndrome
Many people with this syndrome experience speech problems and some of them experience visual loss. In addition, people with disabilities may experience intellectual loss (dementia) and psychiatric symptoms such as behavioral problems, personality changes, and depression [2]. Hallervorden-Spatz syndrome is characterized by an abnormal increase in iron in specific areas of the brain. In people with this syndrome, a sign in their brains is seen as a tiger eye by radiological imaging (MRI), which indicates the accumulation of iron in the brain of these patients [3]. Researchers have described the classic and unusual forms of Hallervorden-Spatz syndrome. The classic form usually appears in early childhood and causes severe movement problems that increase rapidly. Unusual features of the syndrome appear to begin in childhood or adolescence and develop slowly. The signs and symptoms of this form are different, but they are more common than the classic form, which includes speech disorders and psychiatric problems [4].
The Etiology of Hallervorden-Spatz Syndrome
Hallervorden-Spatz syndrome is caused by a mutation in the PANK2 gene located on the short arm of chromosome 20 at 20p13. This gene provides instructions for the synthesis of an enzyme called pantothenate kinase 2 [5] (Figure 1). This enzyme plays a role in mitochondria, the centers of energy production within cells, and plays an important role in the formation of a molecule called coenzyme A. In all living cells, Co-enzyme A is essential for the body to produce energy from carbohydrates, fats and some protein building blocks (amino acids) [6]. Mutation in the PANK2 gene may result in abnormal production of the pantothenate kinase 2 or no production of this enzyme.
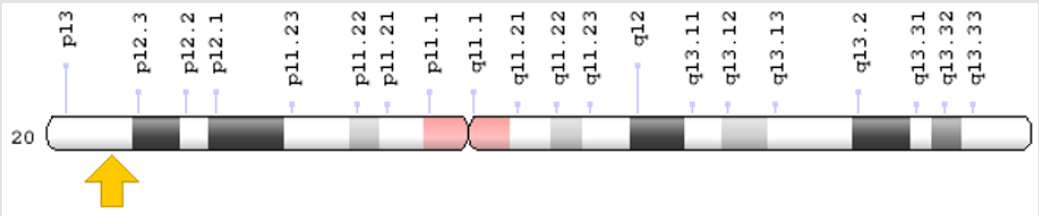
Figure 1: Schematic overview of chromosome 20 where the PANK2 gene is located in the short arm of chromosome 20p13.
Functional pantothenate kinase 2 deficiency disrupts coenzyme A production and creates potentially harmful compounds in the brain. This construction leads to swelling and tissue damage and accumulates iron in some parts of the brain abnormally. Researchers have not determined how these changes lead to specific features of the Hallervorden-Spatz syndrome. Because pantothenate kinase 2 works in the mitochondria, signs and symptoms of the disease may be related to reduced energy production [7]. Hallervorden-Spatz syndrome follows an autosomal recessive inheritance pattern. Therefore, two copies of the mutated PANK2 gene (one from the father and the other from the mother) are required to cause the syndrome, and the chance of having a child with autosomal recessive syndrome is 25% for each possible pregnancy [7] (Figure 2).
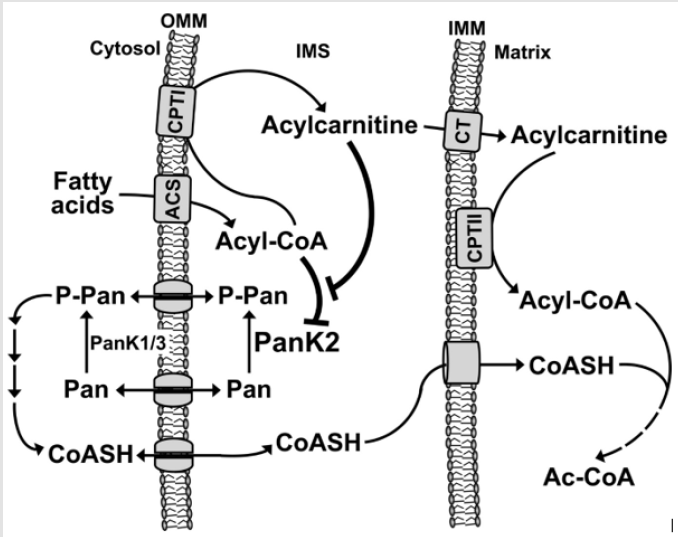
Figure 2: Schematic of the biochemical pathway of the PANK2 gene.
Frequency of Hallervorden-Spatz Syndrome
Hallervorden-Spatz syndrome is a very rare genetic disorder with an estimated prevalence of about 1 in 3 million people worldwide [8].
Diagnosis of Hallervorden-Spatz Syndrome
Hallervorden-Spatz syndrome is diagnosed based on clinical and clinical findings of patients and some pathological tests. The most accurate method for detecting this syndrome is molecular genetic testing of the PANK2 gene to investigate the presence of possible mutations [8] (Figure 3).
Therapeutic Pathways of Hallervorden-Spatz Syndrome
The strategy of treatment and management of Hallervorden- Spatz syndrome is symptomatic and supportive. Treatment may be coordinated with a team of specialists including a neurologist, an orthopedist, a physician, a psychiatrist, and other health care professionals. There is no effective treatment for this syndrome and all clinical measures are to reduce the suffering of patients. Genetic counseling is also needed for all parents who want a healthy child [9] (Figure 4).

Figure 3: Radiologic Imaging of Brain Disorders with Hallervorden-Spatz Syndrome.
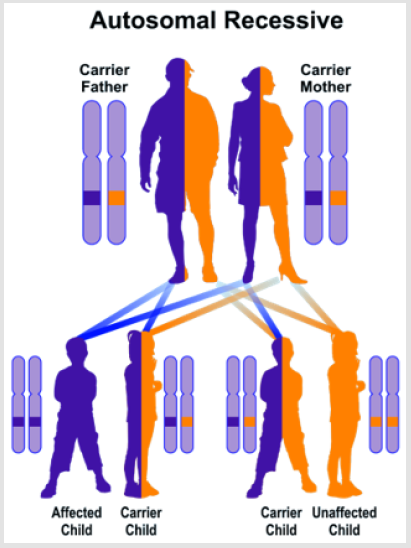
Figure 4: Schematic overview of the autosomal recessive inheritance pattern, which also follows the Hallervorden-Spatz syndrome.
History of Hallervorden-Spatz Syndrome
Hallervorden-Spatz syndrome was first reported in 1922 by Hallervorden and Spatz [9].
Discussion and Conclusion
PKAN is inherited as an autosomal recessive genetic condition and is described as being classical or atypical. Classic PKAN typically appears in early childhood with symptoms that worsen rapidly. Atypical PKAN, which progresses more slowly, appears later in childhood or early adolescence. Some people have been diagnosed in infancy or adulthood, and some of those affected have characteristics that are between the two categories. In cases of neurodegeneration with brain iron accumulation (NBIA) that are not caused by PKAN, the movement-related symptoms (such as dystonia) may be very similar. Nine additional genes causing various subtypes of NBIA have been identified at this time. For those without a specific diagnosis or known cause of NBIA, symptoms are more varied because there are probably several different causes of neurodegeneration in this group. There is a subgroup of patients with moderate to severe intellectual disability.
Also, seizure disorders are more common among non-PKAN individuals. There is no specific treatment for individuals with PKAN. Treatment is directed towards the specific symptoms that appear in each individual. Research is focusing on a better understanding of the underlying cause of this disorder, which may eventually help to find a more comprehensive treatment. Drugs that reduce the levels of iron in the body (iron chelation) have been attempted to treat individuals with PKAN. These early agents were proven ineffective and can cause anemia. A clinical trial of the drug deferiprone was completed for PKAN and results were published in 2019. The results suggested a possible modest slowing of disease progression, although the statistical analysis of the data was not able to prove this as significant [10].
References
- Gregory A, Hayflick SJ (2005) Neurodegeneration with brain iron accumulation. Folia Neuropathol 43(4): 286-96.
- Gregory A, Hayflick SJ (2017) Pantothenate Kinase-Associated Neurodegeneration. 2002 Aug 13. In Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A (Eds.). GeneReviews®. Seattle (WA), University of Washington, Seattle, Washington, USA, 1993-2017.
- Hartig MB, Hörtnagel K, Garavaglia B, Zorzi G, Kmiec T, et al. (2006) Genotypic and phenotypic spectrum of PANK2 mutations in patients with neurodegeneration with brain iron accumulation. Ann Neurol 59(2): 248-256.
- Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, et al. (2003) Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med 348(1): 33-40.
- Hayflick SJ (2003) Pantothenate kinase-associated neurodegeneration (formerly Hallervorden-Spatz syndrome). J Neurol Sci 207(1-2): 106-107.
- Hayflick SJ (2003) Unraveling the Hallervorden-Spatz syndrome: pantothenate kinase-associated neurodegeneration is the name. Curr Opin Pediatr 15(6): 572-577.
- Houlden H, Lincoln S, Farrer M, Cleland PG, Hardy J, et al. (2003) Compound heterozygous PANK2 mutations confirm HARP and Hallervorden-Spatz syndromes are allelic. Neurology 61(10): 1423-1426.
- Pellecchia MT, Valente EM, Cif L, Salvi S, Albanese A, et al. (2005) The diverse phenotype and genotype of pantothenate kinase-associated neurodegeneration. Neurology 64(10): 1810-1812.
- Shevell M (2003) Hallervorden and history. N Engl J Med 348: 1725-1726.
- Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, et al. (2001) A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet 28(4): 345-349.
