 Case Report
Case ReportAbstract
We described a typical clinical case where the simply quick look opens the way for you. We think we must always remember that anything that does not fit perfectly into normality can be pathological and therefore we must not underestimate it. We believe that this clinical case can be very useful for students or young doctors because if we do not know how to give explanations in medicine, at least you should ask a second opinion to other colleagues.
Case Presentation
An apparent 49-years-old healthy man accompanied his son of 9 years old to Emergency Department because of fever and hyperchromic urine from two days. When I saw this father, I am stay stunned. After an accurate clinical examination, I concluded that the child could have an Epstein-Barr virus infection. He had splenomegaly, hypertransaminasemia, pharingeal purulent exudate and the rapid test for EBV-IgM resulted positive. But I could not look away from the father: the body of evidence was too substantial to disregard, and the light bulb had turned on!
Investigation
Aware of these evidences, I asked to father if his skin color is always been so dark (Figure 1a). He responded me that he noted some changes in his color skin from about two years. I asked if he had any particular symptoms and he answered that he had a period about four months ago with extreme asthenia associated with poliarthralgia, but they are now a distant memory. When he showed me his mouth (Figures 1b & 1c) my suspicious further increased, and I recommended him to perform some blood exams which definitively confirmed the diagnosis of Addison’s disease (ACTH levels: 975 pg/ml and hypo-cortisolemia: 1.2 μg/dl).

Figure 1a: Hyperpigmentation of patient’s face.
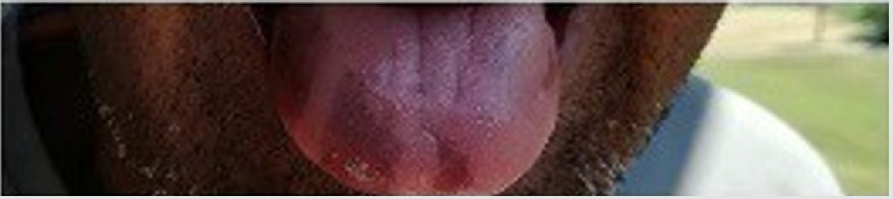
Figure 1b: Melanodermic areas of the tongue.
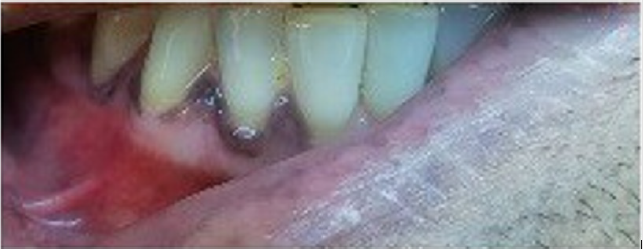
Figure 1c: Melanodermic gengivities.
Treatment
We immediately recommended an endocrinologic examination and the endocrinologist, after confirming the diagnosis of Addison’s disease, started therapy with cortisone acetate and set the appropriate endocrinologic follow-up.
Outcome and Follow-Up
Analyzing the past history of the patient, he reported an apparent dysphagia for some foods and a weight loss of about 5 Kg in the last 5 months. A few months earlier, he contacted both his family doctor and odontologist and did explain them his symptoms, but they did not give great importance to these apparently nuanced clinical disorders. The next abdominal TAC showed only an hypoplastic aspect of adrenal glands, confirming the autoimmune etiology of Addison’s disease. The second level blood tests showed the glucocorticoid involvement only and a mild hypo- thyroidism. The patient just after few weeks of replacement therapy, reported a great improvement in his general conditions especially the desire to work is back and all the previously unexplained symptoms such as mild anxiety, depression and apathy are now a distant memory.
Discussion
Addison’s disease (AD) or primary adrenocortical insufficiency is a rare autoimmune pathology characterized by inability of the adrenal cortex to produce sufficient amounts of glucocorticoids and mineralocorticoids [1]. The disease often begins between the 2nd and 4th decade of life and affects women more often than men. At the time of diagnosis, and before the start of treatment, the patients can be critically ill with increased risk of death because of a stressful event which may eventually precipitate acute adrenal insufficiency [2]. On the contrary, once the disease is diagnosed and treated with hormone replacement therapy [3], life expectancy has been considered normal [4]. The symptoms are often characterized by fatigue, nausea, dizziness and weight loss, sometimes by salt-craving but the pathognomonic signs are hyperpigmentation of the skin.
The elevated plasma ACTH as well as low serum cortisol levels serves as diagnostic criteria for Addison’s disease [5]. Although tuberculosis was the predominant cause of AD earlier, autoimmunity, often not isolated1, is now responsible for most of the cases in developed countries [5]. Despite continuous loss of adrenocortical cells, AD may remain subclinical for long periods before overt disease develops [2]. Sometimes skin and mucosal hyperpigmentation, albeit evident, can remain misdiagnosed if the disease is not well known and therefore, we do not think about it. Analyzing the familial clinical history, the patient’s mother suffers from rheumatoid arthritis and the brother from type-1 diabetes mellitus. In conclusion, when you think that the patient may have a pathological aspect and you do not know why, do not underestimate him but ask a second opinion to other colleagues.
Author’s Statement
MM made the diagnosis and write the manuscript together with MP who began the therapy. GG searched literature data. All authors approved the final version of the manuscript.
Additional Contributions
We thank the patient for providing permission to share his information
Conflict of Interest Disclosures
All authors declare no conflict of interest.
References
- Erichsen M, Løvas K, Skinningsrud B, Wolff AB, Undlien DE, et al. (2009) Clinical, Immunological, and Genetic features of Autoimmune Primary Adrenal Insufficiency: Observations from a Norwegian registry. J Clin Endocrinol Metab 94(12): 4882-4890.
- Hellesen A, Bratland E, Husebye ES (2018) Autoimmune Addison’s disease-An update on pathogenesis. Annals of Endocrinology (Annales D'Endocrinologie, English Edition) 79(3): 157-163.
- Bornstein SR, Allolio B, Arlt W, Barthel A, Don Wauchope A, et al. (2016) Diagnosis and treatment of primary adrenal insufficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 101(2): 364-389.
- Erichsen MM, Løvås K, Fougner KJ, Svartberg J, Hauge ER, et al. (2009) Normal overall mortality rate in Addison’s disease, but young patients are at risk of premature death. European Journal of Endocrinology 160(2): 233-237.
- Hahner S, Christina Spinnler, Martin Fassnacht, Stephanie Burger Stritt, Katharina Lang, et al. (2015) High incidence of adrenal crisis in educated patients with chronic adrenal insufficiency: a prospective study. J Clin Endocrinol Metab 100(2): 407-416.
