 Review Article
Review ArticleAbstract
Most cancer types are characterized by dysregulated epigenomes at multiple regulatory layers including methylome, histone modifications, chromatin accessibility and chromosome conformation. Previous research on these epigenomic features has been performed using the bulk analysis of cells (pools of millions of cells), therefore preventing the study of cell-to-cell heterogeneity at these regulatory levels, which are extensively reprogrammed in cancer. Recent-developed methods enable the study of epigenomes at a single-cell resolution. Using these novel tools and algorithms, cancer researchers can now study cancer epigenomes at an unprecedented detail and in parallel with single-cell genomic, transcriptomic and proteomic analyses. Considering the presence of heterogenous cell subpopulations in a tumor, it is crucial to study cancer epigenomes at a single-cell level to better understand cancer epigenetics and ultimately to develop highly effective treatment strategies against cancer. Here, we review recent single cell epigenomics methods including several multi-omics approaches, with a particular focus on their applications in cancer research.
Keywords: Single Cell; Epigenomics; Cancer; Epigenetics; Sequencing
Abbreviations: scRRBS: Single-Cell Reduced Representation Bisulfite Sequencing; scBS-seq / scPBAT: Single-Cell Bisulfite Sequencing / Single-Cell Post-Bisulfite Adaptor Tagging; scWGBS: Single-Cell Whole Genome Bisulfite Sequencing; scCHIP-seq: Single- Cell Chromatin Immunoprecipitation followed by Sequencing; scATAC-seq: Single-Cell Assay for Transposase-Accessible Chromatin using sequencing; scDNase-seq: Single-Cell DNase sequencing; scMT-seq: Single-Cell Methylome and Transcriptome Sequencing; scTrio-seq: Single-Cell Triple Omics Sequencing; sc-GEM: Single-Cell Analysis of Genotype, Expression and Methylation; scCOOL-seq: Single-Cell Chromatin Overall Omic-scale Landscape Sequencing; scNOMe-seq: Single-Cell Nucleosome Occupancy and Methylome-Sequencing; scNMT-Seq: Single-cell Nucleosome; Methylation and Transcription Sequencing
Introduction
A single tumor mass is comprised of many different subpopulations of cells. During evolutionary trajectory of a tumor, cells with different molecular features are likely to evolve and interactions between these heterogenous cell subpopulations in tumor are highly dynamic [1]. This intratumoral heterogeneity (ITH) is regulated in multiple levels and each cell subpopulation in a particular tumor can have distinct genomic, epigenomic, transcriptomic and spatial characteristics, effecting their metastatic potential and chemoresistance [2]. How the complex contributions of each subgroup in a complete population of cells in tumor affects the clinical progression of the disease is not completely understood. This is mostly due to the fact that cancer research previously has been limited to the analysis of bulk cells without dissecting heterogenous cell populations into subgroups with similar molecular features, therefore these studies have only reflected average profile of complex subgroups of cells, called subclones. Tumors are characterized by dysregulated epigenomes. Epigenetic modifications contribute to tumor cell heterogeneity resulting in, for example, differential responses to targeted therapies [3].
Epigenetic regulation encompasses many biological layers including chemical modifications to genomic DNA (for example, methylation of cytosines at CpG dinucleotides), chromatin conformation and accessibility, post-translational modifications of histones and transcription factor (TF) binding dynamics. Since epigenetic misprogramming is highly variable between subpopulations of tumor cells in all these layers of regulation, epigenomic profiling of cancer cells and also non-cancer cells present in the tumor microenvironment, at a single-cell resolution is essential in cancer research. Recent advances in single-cell technologies have enabled the characterization of cellular heterogeneity in mixed cell populations such as in cancer and direct analysis of molecular mechanisms in a single-cell. Cancer scientists now have the tools to study cancer at a single-cell resolution, with new methods and computational algorithms to analyse the single-cell data, which are still rapidly emerging in the field. In addition to methods with which single-cell genome and single-cell transcriptome sequencing can be performed, methods to profile epigenome of a single-cell have also been developed and continue to be improved. With these single cell epigenomics tools, it is now possible to study a single cancer cell in terms of all forms of epigenetic regulation (for example, DNA methylation, chromatin accessibility, histone modifications and 3D chromatin topology). In this article, we will review recent single cell epigenomics methods with a particular focus on their applications in cancer research.
DNA Methylome Analysis at Single-Cell Resolution
In cancer, DNA methylation aberrancies is highly evident and it is considered as a hallmark of human cancers [4]. These epigenomic alterations, for instance, can result in silencing of tumor suppressors due promoter hypermethylation or in upregulation of oncogenes due to gene-body hypermethylation. The regulation of DNA methylome is highly complex in cancer and this topic was reviewed elsewhere [4-6]. Here, we touch upon some single-cell methods developed in recent years, which enable us to study methylomes of single cancer cells.
scRRBS (Single-Cell Reduced Representation Bisulfite Sequencing)
The first single-cell methylome analysis was reported in 2013 [7]. In this study, authors modified the original bulk RRBS method [8] and entire protocol was performed in a single tube prior to bisulfite conversion, minimizing DNA losses arising from multiple purification steps. In bisulfite conversion, unmethylated cytosines are converted to uracils due to deamination events, whereas methylated cytosines remain unconverted, thus allowing to study DNA methylation at single-base resolution after generating sequencing libraries of bisulfite-converted DNA template. In this method, unmethylated lambda DNA was used as spike-in to control for the false-positives which are due to non-conversion of unmethylated cytosines. It should be noted that this method does not discriminate between 5-methylcytosine (5-mC) and less frequent 5-hydroxymethylcytosine (5-hmC). Recently, Pastore et al. showed the presence of intratumoral epigenetic diversity in chronic lymphocytic leukemia (CLL) by comparing bulk RRBS and multiplexed scRRBS results [9]. They demonstrated that the correlation between DNA methylation and transcription is lower in CLL cells when compared to the same correlation for normal B cells in bulk analyses, however they found a higher correlation between DNA methylation and transcription in CLL cells when analyses were performed in single cells, reflecting epigenetic diversity in CLL cells. They concluded that this epigenetic heterogeneity in leukemic cells may facilitate the emergence of novel cell states with a possibly higher fitness potential, resulting in, for example, higher resistance to therapy.
scBS-seq / scPBAT (Single-Cell Bisulfite Sequencing / Single-Cell Post-Bisulfite Adaptor Tagging)
This protocol uses a modification of post-bisulfite adaptor tagging (PBAT) used in bulk samples and it minimizes DNA loss from single cells [10,11]. In this method, sequencing adaptors are ligated to fragmented DNA after bisulfite conversion unlike the other common BS-seq methods (such as scRRBS) where adaptor tagging was performed before bisulfite treatment which results in simultaneous DNA fragmentation and degradation. With this technique, higher recovery rates (around 5-fold more CpGs and 1.5-fold more CpG islands) can be achieved at equivalent sequencing depth when compared to scRBBS method. The functional consequences of epigenomic diversity in cancer remain largely unknown since bulk DNA or RNA analyses are inadequate for this particular purpose. One potential future use of scBS-seq technique can be the analysis of epigenetic plurality of cancer cell states in a tumor mass, in terms of their methylome. This epigenetic heterogeneity might explain why some cancer cells metastasize to distal sites in the body and/or resist to chemotherapy, whereas some do not.
scWGBS (Single-Cell Whole Genome Bisulfite Sequencing)
Similar to scBS-seq, this technique also takes advantage of postbisulfite adaptor ligation protocol with which relatively low amount of DNA is lost [12]. This method does not focus on CpG islands as scRBBS method does; but rather covers CpGs in a cumulative manner. In contrast to scBS-seq method, library complexity in scWGBS is relatively low, because this method does not require any pre-amplification step. Gkountela et al. applied this method to single tumor cells that are dissociated from circulating tumor cell (CTC) clusters [13]. They show that CTC cluster dissociation into single constituent cells leads to DNA methylation remodelling at some critical loci (binding sites for OCT4, SOX2, NANOG, and SIN3A) in these single circulating tumor cells. This study highlights the importance of single cell methylome research in providing essential information on epigenetic dynamics of single circulating tumor cells which pose an increased risk for metastasis when they form multicellular clusters. There are multiple other methods developed in recent years which allow studying DNA methylation profiles in single cells [14-16].
In one study, Li et al. used single ovarian cancer cells isolated from formalin-fixed and paraffin-embedded (FFPE) human ovarian cancer tissue using laser capture microdissection to explore DNA methylome heterogeneity between these single cells from the same subpopulation of tumor tissue [15]. We anticipate that many other single-cell DNA methylation analysis methods will be developed, and existing tools will be improved in the near future, reducing the time and cost associated with these techniques. Also, it is very likely that number of cancer studies reporting the use of these protocols will exponentially increase, providing novel insights in the understanding of methylome regulation in cancer, which were previously missed due to bulk analyses. It is also worth noting that DNA modifications other than 5-cytosine methylation (such as 5-formylcytosine, 5-hydroxymethylcytosine and 5-carboxylcytosine) can be measured in a single-cell level using recently developed techniques [17-19]. Thus, these relatively low abundance DNA modifications which form multiple epigenetic layers of molecular connectivity between genome and its functional output can be studied in single cancer cells, providing a great multitude of new data.
Single-Cell Histone Modification Analysis
scCHIP-seq (Single-Cell Chromatin Immunoprecipitation followed by Sequencing)
Bulk CHIP-seq is used in cancer research to map histone modifications and protein-DNA interaction (such as TF binding sites) genome-wide. However, the limitation of this method is that it yields averaged epigenetic profiles of all cells in an analysed population, masking any histone modification and protein-DNA interaction heterogeneity between single-cells in this population of cells. To overcome this obstacle, single-cell modification of this method was developed with the use of microfluidics and DNA barcoding [20]. With the availability of this method, the heterogeneity of key histone marks such as H3K4me3 which represent active transcriptional state, or H3K27me3 which is associated with repressed transcription can be measured in a population of cells in a tumor mass. Therefore, multiple epigenetic dimensions with different functional outcomes (transcriptional activation / repression) can be profiled simultaneously at a singlecell resolution, providing more detailed picture of epigenetic regulation in cancer.
The identification of heterogeneous stromal cell populations in the tumor microenvironment was previously hampered by bulk omics analyses. scCHIP-seq can be performed to answer questions related to the precise cellular composition of the tumor microenvironment, for instance, how variable histone modifications between stromal cells present in close proximity to cancer cells are, and how this epigenetic heterogeneity affects their coevolution with surrounding cancer cells. Considering the role of stromal cells in promoting multiple processes in cancer such as neoangiogenesis, drug resistance, extracellular matrix (ECM) remodelling, metastasis and immune evasion, it is of high importance to study stromal cells at a single-cell resolution to provide new therapeutic strategies [21]. Another recently developed single-cell method which allow the profiling of histone modifications is CUT&Tag (Cleavage Under Targets and Tagmentation) [22]. It also maps transcription factor (TF) binding and accessible DNA in parallel, similar to multi-omics approaches that we will mention below. In this study, they reported that chromatin profiling is sufficient to discriminate single cell types.
Single-Cell Chromatin Accessibility and Chromosome Conformation Analysis
scATAC-seq (Single-Cell Assay for Transposase- Accessible Chromatin using Sequencing)
This method is used to identify active regulatory regions of genome characterized by lower density of nucleosomes (therefore, accessible sites of genome) in single cells [23,24]. It uses prokaryotic Tn5 transposase to insert sequencing adaptors to nucleosome-free elements of the genome where presence of nucleosomes does not block its activity. scATAC-seq allows mapping the accessible genome of individual cells with the use of microfluidic devices, enabling the study of epigenetic regulatory variation between these cells. In one study, the authors first created a mixed cell population using two oesophageal adenocarcinoma cell lines and one non-cancer cell line in equal proportions [25]. Using scATAC-seq data and an algorithm which they developed (named Scasat, single-cell ATACseq analysis tool), they were able to cluster these heterogenous cell population based on chromatin accessibility information. Similar approach can be used to identify, for example, epigenetically diverse subpopulations in a tumor mass, allowing better characterization of molecular mechanisms in subgroups of cells present in the same microenvironment.
Using scATAC-seq, Corces et al. were able to show the presence of regulatory heterogeneity in the epigenome of acute myeloid leukemia (AML) cells at distinct time points during cancer evolution [26]. As in this study, the analysis of chromatin accessibility data combined with functional cell states in individual cells enables direct characterization of the interaction between these two layers. In terms of cancer research, being able to study the relationship between chromatin accessibility patterns and disease progression with methods such as scATAC-seq opens up new research directions with exciting potential outcomes. Satpathy et al. applied scATAC-seq to basal cell carcinoma cells to identify regulatory landscapes of tumor, stromal and immune cell types in the tumor microenvironment [27]. They also performed the method on tumor biopsies before and after PD-1 blockade to characterize chromatin regulators of intratumoral T-cell exhaustion after checkpoint blockade. These findings may help to develop therapeutic interventions which synergize with PD-1 blockade in cancer treatment. There have been some recent improvements on scATAC-seq method which enable single-cell epigenomic profiling at a massive scale such as dsciATAC-seq (droplet-based single-cell combinatorial indexing for ATAC-seq) [28]. These techniques allow high-throughput analysis of single-cell epigenomes, providing new opportunities for single-cell cancer research.
scDNase-seq (Single-Cell DNase Sequencing)
Similar to scATAC-seq, scDNase-seq can be used to map active regulatory regions in the genome with low nucleosome presence (therefore, sensitive to DNase-I cleavage due to reduced protection from nucleosomes) in single cells [29]. These DNase-I hypersensitive sites (DHS) reflect chromatin accessibility patterns and activity of regulatory genomic elements, thus providing important data on the epigenome of a single-cell. In the same study where they reported the development of scDNase-seq, the authors applied this technique to follicular thyroid carcinoma (FTC) cells dissected from formalinfixed paraffin-embedded (FFPE) slides and they identified more than thousand tumor-specific DNase I hypersensitive sites (DHS) in these single cells. Analysis of FTC samples from three different patients revealed that very few DHSs were shared between three samples, showing the patient specificity of most DHSs identified. It can be inferred from this data that tumours in different patients may progress with different underlying epigenetic mechanisms.
scHi-C
Besides chromatin accessibility, chromosomal architectures and spatial nuclear rearrangement provide important information on the epigenetic regulation of gene expression. In contrast to ensemble Hi-C where average chromosome conformations of millions of cells are analysed, in single-cell Hi-C, genome-wide chromosomal organization in a single nucleus is studied [30,31]. In this method, after restriction enzyme (RE) digestion following DNA crosslinking, chromatin fragments are ligated based on their spatial proximity in the nucleus. Sequencing and analysis of these proximity-dependent ligation products provide useful information on topological domain organization of chromatin and long-range regulatory interactions which may show cell-to-cell variability. One can imagine the potential application of this method in characterizing heterogeneity in chromosomal conformation within cancer cells which are seemingly identical, but phenotypically diverse. Analyses at this epigenetic layer will provide insights into functional consequences of topologically associating domains (TADs) in single cancer cells.
scHi-C will also be valuable in studying the spatial organization of chromosomes in rare cell types which cannot be obtained in sufficient numbers for conventional Hi-C experiments. For example, epigenomes of rare cell types such as circulating tumor cells (CTC) and disseminated tumor cells (DTC) can be profiled with scHi-C (and also with any other single-cell epigenomic method), thus allowing better identification of epigenetic control mechanisms in these cells. In addition to the identification of topologically associated domains (TADs) in single cells with scHi-C, lamina-associated domains (LADs) can also be mapped genome-wide in single cells using another approach [32]. In this study, they reported the inverse link between gene activity and lamina contacting regions, and the positive correlation of these regions with a heterochromatin mark, H3K9me3, in single cells.
Single-Cell Multi-Omics Approaches
Analysis of only one molecular feature (such as only methylome) from a single-cell provides limited and incomplete information, because a cellular state is determined by the highly complex interplay of multiple molecular layers in a cell. Therefore, in addition to single-cell omics approaches mentioned above, several single-cell multi-omics techniques with great applicability potential in cancer research have been developed in recent years. Here, we review single-cell multi-omics approaches which include single-cell epigenomics analyses, and exclude those which provide data only on genomic, transcriptomic and/or proteomic levels.
scMT-seq (Single-Cell Methylome and Transcriptome Sequencing)
This method enables simultaneous profiling of DNA methylome and transcriptome from the same single-cell, providing an opportunity to study the correlation between these two omics data [33]. In scMT-seq, cellular membrane is lysed, however, nuclear membrane is kept intact and isolated by microcapillary picking. mRNA isolated from cytoplasmic component is amplified using a modified version of Smart-seq2 method for transcriptome analysis and nuclear genome is used for methylome profiling in parallel [34,35]. This technique takes advantage of scRRBS method for methylome analysis, whereas a similar method named scM&T uses genome-wide bisulfite sequencing for DNA methylation profiling [36]. Both of these parallel methylome and transcriptome sequencing methods in single cells can help to dissect complex interactions between epigenome and transcriptome, and are powerful tools to study cellular heterogeneity, which is intrinsically strong in cancer, in these different omics layers.
The diversity of drug resistance mechanisms in cancer is not completely known. One potential use of single-cell multi-omics approaches in cancer can be the identification of mechanisms regulating drug insensitivity in both genomic and epigenomic levels. By using methylome and transcriptome data acquired via scMT-seq or scM&T-seq, cancer researchers can compare single chemosensitive and chemoresistance cancer cells of the same type in terms of multiple molecular features and identify (epi) genomic processes driving resistance to therapy. These studies will be invaluable in personalized medicine approaches for cancer treatment. By increasing the resolution and size of data, they will have the potential to provide more effective treatment strategies in cancer.
scTrio-seq (Single-Cell Triple Omics Sequencing)
Hou et al. developed a sequencing technique which allows simultaneous analysis of genome, epigenome and transcriptome at a single-cell resolution [37]. They were able to group 25 single hepatocellular carcinoma (HCC) cells into two subpopulations using unsupervised hierarchical clustering and principal component analysis (PCA) using data from three omics layers individually. Clustering of cells based on copy number variation (CNV) patterns (genomic level), global DNA methylation (epigenomic level) and gene expression differences (transcriptomic level) separately were highly consistent, showing the power of this method to dissect heterogenous cancer cell populations into subpopulations with similar genome, methylome (epigenome) and transcriptome characteristics; which is otherwise impossible with bulk analysis. This study also shows the capability of multi-omics single-cell approaches to decipher inter-omics regulation at a single-cell level (for example, the identification of mutual relationships between epigenome and transcriptome).
A similar approach called sc-GEM (single-cell analysis of Genotype, Expression and Methylation) which identifies methylation patterns at specific loci (not globally as in scTrio-seq) was used to cluster single cells from primary lung adenocarcinomas in different patients and single cells from one non-tumor lung tissue, by using DNA methylation state of a panel of genes known to be aberrantly methylated in this cancer type [38]. They were able to group cells into two clusters based on their DNA methylation profiles. Most of the cells from non-tumor sample and tumor cells with wild-type EGFR status are grouped in one cluster, whereas high proportion of tumor cells with EGFR mutation formed a different cluster. This data indicated that both genomic and epigenomic heterogeneity can be employed to dissect heterogenous cell populations observed in cancer into different clusters using a multi-omics single cell approach.
scCOOL-seq (Single-Cell Chromatin Overall Omic-Scale Landscape Sequencing)
This multi-omics single cell sequencing technique makes the measurement possible from different layers of epigenetic regulation in a single-cell unlike previously mentioned methods where the analysis of only one epigenomic layer is performed at a single-cell resolution [39]. scCOOL-seq is the method of choice if simultaneous profiling of chromatin state, nucleosome positioning, DNA methylation (also, genomic analyses such as copy number variation and/or ploidy profiling) in a single cell is desired depending on biological question at hand. In this work, they applied scCOOL-seq to human early embryos and found drastic chromatin remodelling and DNA methylation reprogramming at different stages of development. Same approach can be easily used to study epigenetic reprogramming in cancer evolution and progression at a single-cell resolution. Their strategy also enabled them to discriminate aneuploid cells in a population of cells. Considering the pervasiveness of aneuploidy in cancer, this technique has potential to be utilized in the study of chromosomal aberrations and instability in cancer research [40].
scNOMe-seq (Single-Cell Nucleosome Occupancy and Methylome-Sequencing)
Another single-cell technology that measures multiple aspects of gene regulation in single cells is scNOMe-seq [41]. This method employs GpC methyltransferase (MTase) which methylates cytosines in GpC (not CpG) dinucleotides in DNA regions with low nucleosome occupancy. When combined with bisulfite sequencing, both GpC sites (methylated in accessible chromatin regions by MTase) and endogenous CpG methylation sites can be simultaneously analysed, allowing profiling of chromatin accessibility and methylome (two layers of epigenomic regulation) in a single cell. The advantage of this method over other single-cell chromatin accessibility profiling methods such as scATAC-seq and scDNase-seq is that the use of GpC methylation for determination of chromatin accessibility helps to better distinguish inaccessible chromatin from missing data which is a common problem in countbased methods. The dysregulation of chromatin remodelling and coordinated disruption in the regulation of methylome are major driving factors in cancer [42,43]. With the use of newly developed single-cell multi-omics techniques such as scNOMe-seq, cancer scientists can now study these epigenetic layers participating in cancer development, progression and maintenance in parallel to provide novel insights in understanding of cancer biology at a single-cell level. This multi-omics approach also makes it possible to identify the mutual relationship between chromatin states and DNA methylation patterns, providing previously impossible information on the interaction of multiple epigenetic mechanisms.
scNMT-seq (Single-Cell Nucleosome, Methylation and Transcription Sequencing)
scNMT-seq combines RNA-seq with chromatin accessibility and methylome profiling performed in the same cell [44]. It is built on scNOMe-seq method with the addition of transcriptome analysis. As in scNOMe-seq, it allows parallel profiling of two epigenomic molecular layers (chromatin accessibility and DNA methylation) at a single-cell, making it possible to study these dependent features simultaneously. Combined transcriptome analysis adds another omics layer and enables assessing dependencies and associations between epigenome and transcriptome, which is critical for a complete understanding of regulatory mechanisms in cancer. In addition, it will be quite important to see if epigenetic heterogeneity within single cancer cells is coupled or uncoupled between these different omics layers. We previously reported that epigenetic silencing of RGS10 and RGS2 genes by HDAC1 (histone deacetylase 1) and DNMT1 (DNA methyltransferase 1) contributes to cisplatin chemoresistance in ovarian cancer [45,46]. The regulation of this and other genes known to have essential roles in resistance to therapy can be studied genome-wide using scNMT-seq at a single-cell resolution, incorporating data obtained in multiple epigenomic regulatory layers. Another possible use of this multiomics approach can be in identifying cancer cell subpopulations of which epigenetic silencing or activation of genes regulating chemoresistance (such as RGS10) is more profound relative to other cancer cell subpopulations in the same microenvironment for which epigenetic control does not contribute much to drug resistance.
sci-CAR
This method allows joint profiling of gene expression and chromatin accessibility in high-throughput since it uses split pool barcoding for thousands of single cells [47]. In this study, they applied sci-CAR to cells from a human lung carcinoma cell line collected at different time points after dexamethasone treatment. They were able to cluster untreated and treated cells based on transcriptome or chromatin accessibility data using unsupervised clustering or t-SNE visualization, showing the potential of this technique in scalable profiling of single-cell molecular phenotypes.
Conclusion
Most research on single-cell omics and multi-omics techniques have been consisted of proof-of-concept studies, and publications reporting the application of these methods in various aspects of cancer research are just beginning to emerge. With rapid advances in the field and reducing costs to perform such studies, increasing number of cancer researchers will employ these techniques in their field of study. Another dimension to include in singlecell epigenomics studies is time. Considering the fact that many epigenetic features have different stabilities in time, studying these omics data in temporal dimension will offer interesting insights in single-cell cancer research, such as the change of epigenetic marks over the course of cancer evolution at a single cell level. Identification of some epigenomic marks which are highly dynamic and short-lived will increase the time resolution in cancer studies and help to offer therapies which are timely administered and more personalized.
Though excluded from this study, single-cell proteomics approaches also provide important findings on tumor individuality and characteristics of the other cells present in tumor microenvironment including immune cells [48]. This type of large-scale studies may be highly useful in precision medicine approaches which target tumor and/or immune cells in the same microenvironment. Another possible outcome of these studies may be more accurate cancer patient classification in the clinic. With the development of more advanced computational algorithms which can process these multidimensional and big data obtained in multiomics approaches, the intricate relationships between all these regulatory layers can be explored. This offers unique perfectives to understand the biology of cancer in more depth (Figure 1).
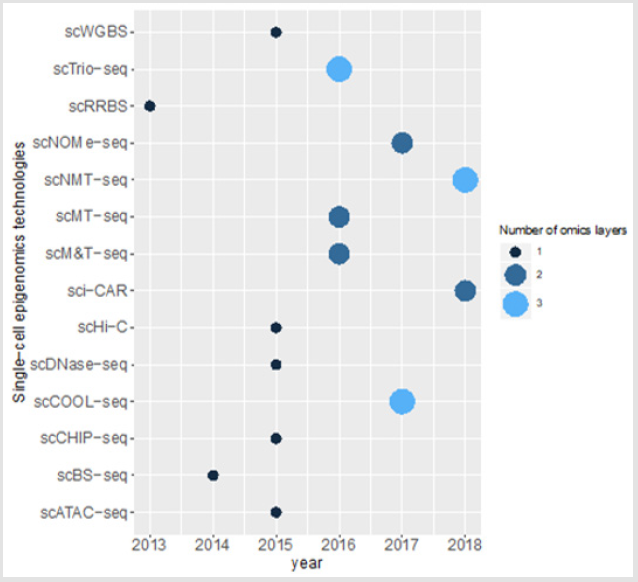
Figure 1: Single cell epigenomics methods and the number of omics layers which can be studied using these technologies.
References
- Tabassum DP, Polyak K (2015) Tumorigenesis: it takes a village. Nat Rev Cancer 15(8): 473-483.
- Hinohara K, Polyak K (2019) Intratumoral Heterogeneity: More Than Just Mutations. Trends Cell Biol 29(7): 569-579.
- Timp W, Feinberg AP (2013) Cancer as a dysregulated epigenome allowing cellulargrowth advantage at the expense of the host. Nat Rev Cancer 13(7): 497-510.
- Liang G, Weisenberger DJ (2017) DNA methylation aberrancies as a guide for surveillance and treatment of human cancers. Epigenetics 12(6): 416-432.
- Pfeifer GP (2018) Defining Driver DNA Methylation Changes in Human Cancer. International journal of molecular sciences 19(4): 1166.
- Michael K, Deborah N, Razi G, Howard C (2016) DNA Methylation in Cancer and Aging. Cancer Res (76) (12): 3446-3450.
- Guo H, Zhu P, Wu X, Li X, Wen L, et al. (2013) Single cell methylome landscapes ofmouse embryonic stem cells and early embryos analyzed using reducedrepresentation bisulfite sequencing. Genome Res 23(12): 2126-2135.
- Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, et al. (2005) Reduced representation bisulfite sequencing for comparative high-resolution DNAmethylation analysis. Nucleic Acids Res 33(18): 5868-5877.
- Pastore A, Gaiti F, Lu SX, Brand RM, Kulm S, et al. (2019) Corrupted coordination of epigenetic modifications leads to diverging chromatin states and transcriptional heterogeneity in CLL. Nat Commun 23 10(1): 1874.
- Clark SJ, Smallwood SA, Lee HJ, Krueger F, Reik W, et al. (2017) Genome-widebase-resolution mapping of DNA methylation in single cells using single-cellbisulfite sequencing (scBS-seq). Nat Protoc 12(3): 534-547.
- Miura F, Enomoto Y, Dairiki R, Ito T (2012) Amplification-free whole-genomebisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res 40(17): e136.
- Farlik M, Sheffield NC, Nuzzo A, Datlinger P, Schönegger A, et al. (2015) Single-cell DNA methylome sequencing and bioinformatic inference of epigenomiccell-state dynamics. Cell Rep 310(8): 1386-1397.
- Gkountela S, Castro Giner F, Szczerba BM, Vetter M, Landin J, et al. (2019) Circulating Tumor Cell Clustering Shapes DNAMethylation to Enable Metastasis Seeding. Cell 176(1): 98-112.
- Mulqueen RM, Pokholok D, Norberg SJ, Torkenczy KA, Fields AJ, et al. (2018) Highlyscalable generation of DNA methylation profiles in single cells. Nat Biotechnol 36(5): 428-431.
- Li Q, Xue X, Li W, Wang Q, Han L, et al. (2017) Heterogeneous DNA methylation status in same-cellsubpopulations of ovarian cancer tissues. Tumour Biol 39(6): 1010428317701650.
- Wang K, Li X, Dong S, Liang J, Mao F, et al. (2015) Q-RRBS: a quantitative reduced representation bisulfite sequencing method forsingle-cell methylome analyses. Epigenetics 10(9): 775-783.
- Zhu C, Gao Y, Guo H, Xia B, Song J, et al. (2017) Single-Cell 5-Formylcytosine Landscapes of Mammalian Early Embryos and ESCs at Single-Base Resolution. Cell Stem Cell 20(5): 720-731.
- Mooijman D, Dey SS, Boisset JC, Crosetto N, van Oudenaarden A (2016) Single-cell5hmC sequencing reveals chromosome-wide cell-to-cell variability and enableslineage reconstruction. Nat Biotechnol 34(8): 852-856.
- Wu X, Inoue A, Suzuki T, Zhang Y (2017) Simultaneous mapping of active DNAdemethylation and sister chromatid exchange in single cells. Genes Dev 31(5): 511-523.
- Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, et al. (2015) Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol 33(11): 1165-1172.
- Zhan HX, Zhou B, Cheng YG, Xu JW, Wang L, et al. (2017) Crosstalk betweenstromal cells and cancer cells in pancreatic cancer: New insights into stromal biology. Cancer Lett 392: 83-93.
- Kaya Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, et al. (2019) CUT&Tag for efficient epigenomic profiling of small samples andsingle cells. Nat Commun 10(1): 1930.
- Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, et al. (2015) Multiplex single cell profiling of chromatinaccessibility by combinatorial cellular indexing. Science 348(6237): 910-914.
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, et al. (2015) Single-cell chromatin accessibility reveals principles ofregulatory variation. Nature 523(7561): 486-490.
- Baker SM, Rogerson C, Hayes A, Sharrocks AD, Rattray M, et al. (2019) Classifying cells with Scasat, a single-cell ATAC-seq analysis tool. Nucleic acids research 47(2): e10.
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, et al. (2016) Lineage-specific andsingle-cell chromatin accessibility charts human hematopoiesis and leukemiaevolution. Nat Genet 48(10): 1193-1203.
- Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, et al. (2019) Massivelyparallel single-cell chromatin landscapes of human immune cell development andintratumoral T cell exhaustion. Nat Biotechnol 37(8): 925-936.
- Lareau CA, Duarte FM, Chew JG, Kartha VK, Burkett ZD, et al. (2019) Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat Biotechnol 37(8): 916-924.
- Jin W, Tang Q, Wan M, Cui K, Zhang Y, et al. (2015) Genome-wide detection of DNase I hypersensitive sitesin single cells and FFPE tissue samples. Nature 528(7580): 142-146.
- Lieberman Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles ofthe human genome. Science 326(5950): 289-293.
- Belton JM, McCord RP, Gibcus JH, Naumova N, Zhan Y, et al. (2012) Hi-C: acomprehensive technique to capture the conformation of genomes. Methods 58(3): 268-276.
- Kind J, Pagie L, de Vries SS, Nahidiazar L, Dey SS, et al. (2015) Genome-wide maps of nuclear lamina interactions in single human cells. Cell 163(1): 134-147.
- Hu Y, Huang K, An Q, Du G, Hu G, et al. (2016) Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol 17: 88.
- Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, et al. (2013) Smart-seq2 for sensitive full-length transcriptome profiling in single cells. NatMethods 10(11): 1096-1098.
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, et al. (2014) Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9(1): 171-181.
- Angermueller C, Clark SJ, Lee HJ, Macaulay IC, Teng MJ, et al. (2016) Parallel single-cellsequencing links transcriptional and epigenetic heterogeneity. Nat Methods 13(3): 229-232.
- Hou Y, Guo H, Cao C, Li X, Hu B, et al. (2016) Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res 26(3): 304-319.
- Cheow LF, Courtois ET, Tan Y, Viswanathan R, Xing Q, et al. (2016) Single-cell multimodal profiling revealscellular epigenetic heterogeneity. Nat Methods 13(10): 833-836.
- Guo F, Li L, Li J, Wu X, Hu B, et al. (2017) Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res 27(8): 967-988.
- Sansregret L, Swanton C (2017) The Role of Aneuploidy in Cancer Evolution. ColdSpring Harb Perspect Med 7(1): a028373.
- Pott S (2017) Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. Elife 6: e23203.
- Kumar R, Li DQ, Müller S, Knapp S (2016) Epigenomic regulation of oncogenesis bychromatin remodeling. Oncogene 35(34): 4423-4436.
- Stirzaker C, Zotenko E, Song JZ, Qu W, Nair SS, et al. (2015) Methylomesequencing in triple-negative breast cancer reveals distinct methylation clusterswith prognostic value. Nat Commun 6:5899.
- Clark SJ, Argelaguet R, Kapourani CA, Stubbs TM, Lee HJ, et al. (2018) scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun 9(1): 781.
- Cacan E, Ali MW, Boyd NH, Hooks SB, Greer SF, et al. (2014) Inhibition of HDAC1 and DNMT1modulate RGS10 expression and decrease ovarian cancer chemoresistance. PLoS One 9(1): e87455.
- Cacan E (2017) Epigenetic regulation of RGS2 (Regulator of G-protein signaling 2) in chemoresistant ovarian cancer cells. J Chemother 29(3): 173-178.
- Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, et al. (2018) Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361(6409): 1380-1385.
- Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, et al. (2019) ASingle-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 177(5): 1330-1345.
