 Short Communication
Short CommunicationAbstract
Genetic variations of microRNAs and their target sites among individuals are increasingly recognized as possible factors underlying various human disorders. Currently, the computer-assisted predictions of microRNA-binding sites rely exclusively on one or few reference genomic sequences and do not take into consideration significant variations within human genomes. There is a need to develop computational approaches to assess the probability of excessively strong or completely disabled microRNA-binding sites upon the known SNP frequencies. We developed the software NEIL to align the individual microRNA and mRNA sequences in the setting of the corresponding genomic SNP map of the targeted gene. This approach allows us to visualize the presence of SNPs within and in the proximity of microRNA-target sites in order to predict the combinatorial effects of SNPs on microRNA-mRNA binding and the expression of the microRNA-regulated genes.
Keywords: MicroRNA; Nucleotide; Variations; Polymorphisms; Alignment; Algorithm
Abbreviations: miR: microRNA; mRNA: messenger RNA; SNP: Single Nucleotide Polymorphism; VTCN1: V-Set Domain Containing T Cell Activation Inhibitor-1; GWAS: Genome Wide Association Studies; CCAS: Case Control Association Studies
Introduction
miRs are evolutionarily conserved, single-stranded, regulatory RNA molecules of about 22 nucleotides long [1]. Deep-sequencing and computational approaches indicate that almost every mammalian mRNA can be targeted by hundreds of miRs, whereas mammalian genomes harbor thousands of miRs, and each miR can potentially influence the expression of hundreds of genes [2]. miRs are involved in various biological processes, such as cell differentiation, organ development, hormonal and neural regulation, immune response, oncogenesis [3,4] and currently viewed as the global regulators of gene expression. Genetic variations of miRs and their target sites among individuals are increasingly recognized as possible factors underlying various human disorders [1,5-7].
The stretch of nucleotides 2-8 from the 5’ end of the miR is called the “seed” region. The Watson–Crick complementarity between the seed region and the target mRNA sequence is considered critical for miR- mediated regulation of target genes [8]. SNPs may either disrupt the miR–mRNA interaction or, in the opposite, create a perfect miR-target site that otherwise is not associated with the given mRNA [1]. Currently, the computer-assisted predictions of microRNA-binding sites rely exclusively on one or few reference genomic sequences and do not take into consideration significant variations within human genomes. There is a need to develop computational approaches to detect and assess the potential impacts of excessively strong or completely disabled miR-binding sites while taking into consideration the various frequencies of specific SNPs [9].
We developed the software NEIL (patent pending) to align the individual miR and mRNA sequences based on Watson-Crick complementarity in the setting of the corresponding genomic SNP map of the targeted gene. Such an approach allows us to visualize the presence of SNPs within and beyond miR-target sites in order to predict the combinatorial effects of SNPs on miR-mRNA binding, and the expression of the miR-regulated genes.
Methods
The NEIL is a string-matching algorithm for the pattern searching. It was designed to recognize the uninterrupted matching of six and more nucleotides of miRs and its mRNA target sequences. The current version of the algorithm distinguishes only the canonical Watson-Crick complementarity (A-T, and G-C). We used TargetScan database as the source of the predicted miRbinding sites on the human MET and VTCN1 mRNAs. The images of the alignments were placed over the images of the corresponding genomic regions of the targeted mRNAs with mapped SNPs. The SNPs with the known Minor Allele Frequency (validated SNPs) can be used to calculate the cumulative probability of weakening or enhancement of miR-mRNA interaction.
Results
The NEIL algorithm precisely recognized the TargetScanpredicted miR-mRNA canonical binding sites and provided for the picturing of the miR-mRNA alignment. When the alignment was superimposed on the genomic map of the appropriate mRNA fragment (the positive strand for human MET, and the negative strand for human VTCN1 genes), the investigators can visualize the location of validated SNPs (within the seed- matching mRNA regions, or just within the miR-matching regions, yet beyond seed-matching areas) and analyze the probability of the possible disruption of miR-target site or, in opposite, the enhancement of miR- mRNA binding (Figure 1).
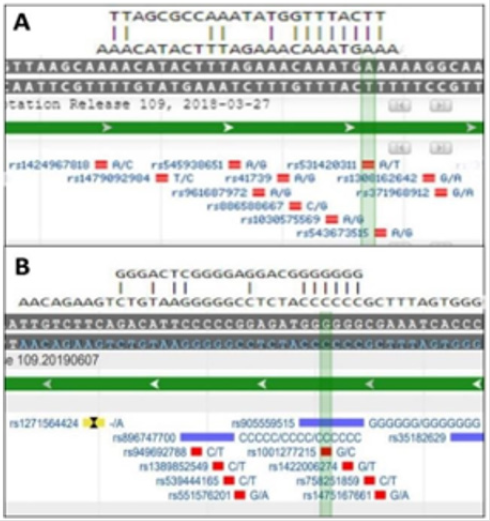
Figure 1: Visualization of miR-mRNA alignments in the
context of the corresponding genomic regions.
A: hsa-miR-579-3p vs human MET mRNA, and B: hsamiR-
328-5p vs human VTCN1 mRNA.
Discussion
Exploration of validated SNPs within miRs and their target regions with the following GWAS and CCAS is a common approach to study the role of genetic susceptibility in the etiology of rare complex diseases, in particular, cancer [6,10,11]. The SNP frequency analysis and identification of SNPs that can increase, decrease, or have neutral effects on miRNA binding are helpful to predict disease-associated SNPs [7,12]. By now, there have been only a limited number of reports on the designed algorithms and databases that assess the possible effects of SNPs on miRs and their target sites. PolymiRTS database contains information about experimentally identified and predicted miR-target sites, polymorphisms in miR-seed regions and links between SNPs in miRNA target sites, expression of quantitative trait loci and results of GWAS [13].
miRNASNP database is aimed to provide information on SNPs in miRs and genes that may impact miR biogenesis and/or miR target binding. The features of this application include expression level and expression correlation of miRs and target genes in different tissues, linking SNPs to the results of GWAS and integrating experimentally validated miR/mRNA interactions. It also includes multiple filters to prioritize functional SNPs [14]. The application allows analyzing only one base difference between the wild type and the SNP-containing mRNA sequences. Our observations, however, indicate the possibility of more than one SNP within the seed- corresponding mRNA regions as well as additional SNPs beyond the seeds [15] (publication in preparation). SNPs nearby miRNA-binding sites that diminish the target accessibility [16] and modify the sequence context outside the target region [17] may also influence the miR-mediated effects of on their genes-targets [18,19].
The NEIL application is designed as a set of webs- available tools to explore the potential combinatorial effects of SNPs within and beyond miR seed- corresponding mRNA sequences and the larger mRNA regions onto which the whole miRs are projected. The approach implies the visualization of miR-mRNA alignment in the context of the genomic map of the corresponding mRNA fragment. The existence and importance of the predicted deviations and disruptions of miR-mRNA binding can be verified in the following genome-wide sequencing experiments as well as GWAS and CCAS.
Acknowledgment
The work was supported by the Faculty Development Grant, Department of Computer Sciences and Department of Biology, Troy University, Alabama, U.S.A.
References
- Chen K, Song F, Calin GA, Wei Q, Hao X, et al. (2008) Polymorphisms in microRNA targets: a gold mine for molecular epidemiology. Carcinogenesis 29(7): 1306-1311.
- Griffiths Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ (2006) miRBase: microRNA sequences, targets and gene nomenclature. Nucleic acids research 34: D140-D144.
- Jin Y, Lee CG (2013) Single Nucleotide Polymorphisms Associated with MicroRNA Regulation. Biomolecules 3(2): 287-302.
- Arnold M, Ellwanger DC, Hartsperger ML, Pfeufer A, Stumpflen V (2012) Cis-acting polymorphisms affect complex traits through modifications of microRNA regulation pathways. PloS one 7(5): e36694.
- Sethupathy P, Collins FS (2008) MicroRNA target site polymorphisms and human disease. Trends in genetics : TIG 24(10): 489-497.
- Landi D, Gemignani F, Barale R, Landi S (2008) A catalog of polymorphisms falling in microRNA-binding regions of cancer genes. DNA and cell biology 27(1): 35-43.
- Saba R, Medina SJ, Booth SA (2014) A functional SNP catalog of overlapping miRNA-binding sites in genes implicated in prion disease and other neurodegenerative disorders. Human mutation 35(10): 1233-1248.
- Lambert NJ, Gu SG, Zahler AM (2011) The conformation of microRNA seed regions in native microRNPs is prearranged for presentation to mRNA targets. Nucleic acids research 39(11): 4827-4835.
- Kofman A (2019) Nucleotide Variations in microRNA-Binding Sites: The Need of Novel Tools for the Nucleic Acid Alignment. Biomedical Journal of Scientific & Technical Research 15(4): 11574-
- Chatterjee N, Chen YH, Luo S, Carroll RJ (2009) Analysis of Case-Control Association Studies: SNPs, Imputation and Haplotypes. Stat Sci 24(4): 489-502.
- Ziebarth JD, Bhattacharya A, Cui Y (2012) Integrative analysis of somatic mutations altering microRNA targeting in cancer genomes. PloS one 7(10): e47137.
- Li W, Li M, Pu X, Guo Y (2017) Distinguishing the disease-associated SNPs based on composition frequency analysis. Interdiscip Sci 9(4): 459-467.
- Ziebarth JD, Bhattacharya A, Chen A, Cui Y (2012) PolymiRTS Database 2.0: linking polymorphisms in microRNA target sites with human diseases and complex traits. Nucleic acids research 40: D216-D221.
- Gong J, Liu C, Liu W, Wu Y, Ma Z, et al. (2015) An update of miRNASNP database for better SNP selection by GWAS data, miRNA expression and online tools. Database (Oxford) 2015: bav029.
- Budmark A, Catalano M, Haley T, Hicks B, Koenen M, et al. (2018) Abstract 490: Single nucleotide variations within and around microRNA-binding sites. Cancer research 78(13).
- Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E (2007) The role of site accessibility in microRNA target recognition. Nature genetics 39: 1278-1284.
- Sun G, Li H, Rossi JJ (2010) Sequence context outside the target region influences the effectiveness of miR-223 target sites in the RhoB 3'UTR. Nucleic acids research 38(1): 239-252.
- Gibson G (2012) Rare and common variants: twenty arguments. Nat Rev Genet 13(2): 135-145.
- Shin C, Nam JW, Farh KK, Chiang HR, Shkumatava A, et al. (2010) Expanding the microRNA targeting code: functional sites with centered pairing. Molecular cell 38(6): 789-802.
