 Mini Review
Mini ReviewIntroduction
It is determined that different venomous terrestrial and marine animals (scorpions, spiders, snakes, centipedes, sea snakes, cone snails, jellyfishes, sea anemones) produce hundreds of biologically active peptides including great number of proteinaceous compounds with large structural and functional diversity [1]. They are used by venomous organisms for protection against their predators and for attacking preys. During the divergent evolution many active peptides belonging to multigene families formed combinatorial libraries representing highly homologous sets of peptide isoforms usually aimed at wide variety of biological targets such as voltagegated sodium and potassium (NaVs and KVs) channels, Transient receptor potential and histamine H1 receptors and many others, all of them are responsible for a physiological and pathophysiological state of an organism, particularly, excitable and non-excitable cells of neuromuscular, neuronal, heart systems etc. [2]. Their dysfunctions and disturbances, as known, result in a number of channelopathies [3]. Many of the venomous peptides being very effective ligands restore the functional activity of different channel subtypes and are currently recognized as pharmacological or therapeutics agents, which are at various stages of preclinical and clinical trials [3].
One of the most studied marine producers of biologically active compounds with a high pharmacological potential is the sea anemones [4,5], including the specimens of Heteractis genus. Two widespread tropical species, the sea anemone Heteractis crispa (formerly Radianthus macrodactylus) and Heteractis magnifica, are known to produce various biologically active peptides such as three protein types of structurally different toxins: the structural type II neurotoxins modulating some NaV channel subtypes [6], APETx-like peptides blocking proton-activated ASICs channels [7] (both with β-defensin like fold), and α-pore-forming toxins (PFTs, named actinoporins with β-strand+α-helix fold) forming pores in cytoplasmic sphingomyelin-containing membranes [8]. Besides, H. crispa and H. magnifica represent the richest source of serine protease inhibitors (having Kunitz fold) and of α-amylase inhibitors (with β-defensin-like fold) [9-13]. The purpose of this minireview is to demonstrate a scientific and pharmacological potential of a huge number of the produced by H. crispa peptides, which biological targets have been shown by now as a wide range of prey’s ion channels, receptors, and serine protease inhibitors.
Heteractis Neurotoxins as Modulators of Voltage Gated Nav Channels
The most studied type of the sea anemone ion channel-targeting ligands has been shown by today to belong to sodium channel type I and II neurotoxins (46-48 aa, respectively) (Figure 1a), which are highly potent modulators of different mammalian NaV subtypes [14]. Their functionally active Arg-loop (Figure 1b) interacts specif ically with so named receptor site-3 of the voltage sensor domain IV [15]. Five neurotoxins of the type II, RTX-I – RTX-V (48 aa, 5 kDa), were isolated from the sea anemone H. crispa in the individual state [6,16-18] (Figure 1a); they are stabilized by three identically localized disulfide bridges, C3-C43, C5-C33, C26-C44 (Figure 1b).

Figure 1: (a) Multiple sequence alignment of the sea anemone toxins class 1a: the neurotoxins of NaV structure type I, ApA (UniProt ID: P01530) and АрВ (P01531) from Anthopleura xanthogrammica, and the neurotoxins of NaV structure type II, RTX-I (P30831) [16], RTX-II (P30783) [17], RTX-III (P30832) [6], RTX-IV (P30784) [18], RTX-V (P30785) [18], and Sh1 (P19651) from Stichodactyla helianthus. Identical and conservative amino acid residues are depicted on a dark and light gray background, respectively. Straight lines show disulfide bridges. Alignment is done using Vector NTI. (b) The superposition of 3D structures theoretical models of the toxins RTX-III (bright blue), ApB (light blue), and the prototype molecule Sh1 (gray, PDB ID: 2Sh1). The models are presented in the form of ribbon diagrams. In the core view, the disulfide bridges and the side chains of Cys and Arg13 residues are visualized using Chimera.3D program.
RTX-I – RTX-V have been shown to be highly toxic for mammalians, their mechanism of an action on different NaV subtypes is currently actively studied in silico and in vitro. We have found that Radianthus neurotoxins modify mammalian NaV 1.1, 1.2, 1.3, 1.6 channel subtypes and insect, BgNaV1 [19]. These toxins significantly slow down the kinetic of a channel inactivation without changing of their activation and prolong a channel action potential, what leads to a massive release of neurotransmitters from the nerve terminals (the data not published). Due to the positive inotropic effect, the neurotoxins can have a cardio stimulating effect, which can be used in a medical practice in a number of extreme situations. Unfortunately, a significant obstruction for using of these toxins is their high toxicity for mammals. Possibly, obtaining of non-toxic mutant analogs will allow overcoming this obstacle and leading to their subsequent clinical use.
Heteractis APETx2-Like Inhibitors of ASICs Channels
The search and study of ASICs channel ligands represent the great interest for pharmacology and neurophysiology also because these channels are involved in a number of pathological processes including neurological and psychiatric diseases [20]. ASIC-targeting ligands can act on the molecular mechanisms of pain generation. They specifically affect the channel subtypes functional activity through their significant potentiating or partial inhibiting [21]. Four new native peptides, π-AnmTX Hcr 1b-1 (Hcr 1b-1) [7,22] and π-AnmTX Hcr 1b-2 (Hcr 1b-2), -3 (Hcr 1b-3), and -4 (Hcr 1b-4) [23] (41 aa, 4.5 kDa), with high sequence identity to APETx2 (42 aa) from Anthopleura elegantissima [24], were obtained from H. crispa (Figure 2a). The peptides are stabilized by three identically localized disulfide bridges, C4–C37, C6–C30, C20–C38 (Figure 2b).
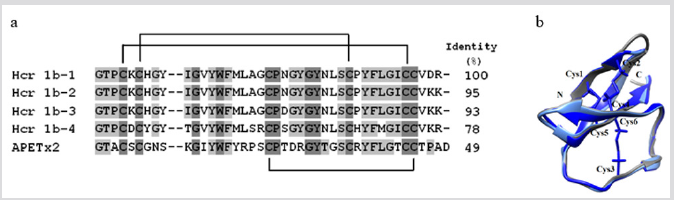
Figure 2: (a) Multiple sequence alignment of amino acid sequences of toxin class 1b: Hcr 1b-1 (UniProt ID: P0DL87) [7], Hcr 1b-2 (C0HL52) [], Hcr 1b-3 (C0HL53), Hcr 1b-4 (C0HL54) from H. crispa [23], and APETx2 (P61542) from A. elegantissima [24]. Identical and conservative amino acid residues are depicted on a dark and light gray background, respectively. Straight lines show three disulfide bridges. Alignment is done using Vector NTI. (b) The superposition of theoretical models of 3D structures of the peptides Hcr 1b-2 (light blue), -4 (dark blue), and the prototype molecule АРЕТх2 (gray, 1WXN). Models are presented in the form of ribbon diagrams. In the core view, the disulfide bridges are visualized using Chimera.3D program.
But while APETx2 reversibly inhibits only homomeric ASIC3 channels (with an IC50 = 63nM) and also some heteromeric channels: ASIC2b+3 (IC50 = 117nM), ASIC1b+3 (IC50 = 0.9μM), and ASIC1a+3 (IC50 = 2μM) by acting at their external side, electrophysiological testing of Heteractis APETx2-like peptides on homomeric ASICs expressed in Xenopus laevis oocytes establishes that they all inhibit not only homomeric ASIC3 channels but also ASIC1a ones [23,25]. The major peptide, Hcr 1b-2, being the first inhibitor of ASIC1a, reversibly inhibits both rASIC1a (IC50 = 4.8μM) and rASIC3 (IC50 = 15.9μM). “The maximum inhibition of these channels by Hcr 1b-2 reaches 64% and 81%, respectively, at saturating peptide concentrations.” Like Hcr 1b-1 [7], Hcr 1b-2 was also shown to exhibit an analgesic effect in the model of acidinduced muscle pain and to reduce significantly the pain threshold of experimental animals [23].
Multigene PFTs of H. crispa and their Biological Template
Native pore-forming toxins (α-PFT) or actinoporins (20kDa) previously obtained from the whole body of H. crispa and Oulactis orientalis (Figure 3a) [8] along with the peptide toxins described above have an important scientific potential because they have the unusual spatial structure [26] (Figure 3b), which stipulates the formation of functionally active protein-lipid pores due to an evolutionary tuned mechanism of interaction with biological targets, cytoplasmic membranes (Figure 3c) [27]. The duality of the nature of actinoporins interaction with targets allows researchers to use them as molecular tools for studying of the cytoplasmic membranes topology and mechanisms of functioning [28]. Recent years successful attempts have been made to create therapeutic agents with anti-tumor, antimicrobial, anti-parasitic activity on the basis of actinoporin structures [29-31]. We have established the presence of H. crispa actinoporin multigene family consisting of 47 representatives [32]. It was shown that the cytotoxic (hemolytic) activity of several native and recombinant members derived from the DNA sequences correlates with the hydrophobicity of their amphiphilic N-termini (28 aa) and the direction of their dipole moments [32].
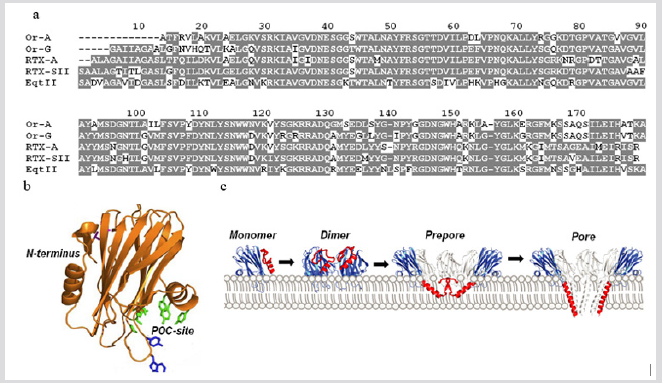
Figure 3: (a) Multiple sequence alignment of H. crispa and O. orientalis actinoporins. Identical and conservative amino acid residues are depicted on a gray background. Alignment is done using Vector NTI. (b) The 3D structure of theoretical model of actinoporin FraC (the pore structure PDB ID: 4TSY). The model is presented in the form of ribbon diagram. (c) The model of FraC pore-formation: an actinoporin monomer binds a membrane and promotes protein-protein interaction between two ones producing the formation of a dimer; then, as a result of dimer interaction with monomers or dimer(s) and due to the insertion of extended actinoporin N-ends in the lipid bilayer, an prepare is first formed, and then a functional pore is created [28].
At the same time, actinoporin binding affinity to biological targets, cytoplasmic membranes, is assigned not only by the interaction of actinoporin POC-binding site with membrane sphingomyelin and phosphatidylcholine, but also by alternative ones of the actinoporin RGD-motive with membrane integrins [33]. Earlier we showed that the cytotoxic effect (IC50) of native RTX-A against the cervix carcinoma cells (HeLa) = 2.26nM, the monocytic leukemia cells (THP-1) = 1.11nM, the breast cancer cells (MDAMB- 231) = 4.64nM, the colon cancer cells (SNU-C4) = 4.66nM, and the promyelocytic leukemia cells (HL-60) = 1.06nM. This actinoporin was demonstrated to exhibit a potential cancer-preventive effect at non-cytotoxic concentrations. These effects were found to exist due to the induction of p53-independent apoptosis as well to the inhibition of oncogenic nuclear factors AP-1 and NF-κB activity [34]. This indicates the possibility of sea anemone PFTs use as targeted tools for specific interaction with the membranes of tumor cells and their destruction. The complex of currently obtained data concerning the possible using of actinoporins as pharmacophores is considered in the works [29-31].
Sea Anemone Kunitz-Type Peptides as Blockers of TRPV1
Today vanilloid receptor TRPV1 is known as a key player of many important physiological processes occurring in almost all organs and tissues. Being the sensors of temperature, mechanical and chemical stimuli (including noxious ones), this receptor is involved in the development of different pain states, which, as rule, accompanied by inflammatory pain associated with diseases of cardiovascular, gastrointestinal, urinary, respiratory, central nervous systems [35]. In this connection, the search and study of action mechanisms of the TRPV1 modulators (both agonists and antagonists) and their alternative role as anti-inflammatory agents, in addition to analgesic one, represent one of the most important tasks of life sciences [36]. To date, most known TRPV1-targeting agonists and antagonists are the low-molecular compounds, many of which undergo, despite the hyperthermia effect, preclinical and clinical trials [37].
But in the past decade some peptides antagonists were found, and they may be regarded as potential therapeutics without this site effect [38-41]. So, the sea anemones H. crispa were shown as a rich source of many serine protease inhibitors of Kunitz-type, some of which had been isolated in the individual state (56 aa, near 6kDa) [10,11,42,43] and more than 70 ones had been deduced from DNA sequences [9] (Figure 4a). The inhibitors are stabilized by three identically localized disulfide bridges, C4-C54, C13-C37, C29-C50 (Figure 4b). They have high sequence similarity and belong to a multigene superfamily forming a combinatorial library of H. crispa Kunitztype peptides [9,44], among which the multifunctional peptides (APHC1, APHC2, APHC3, HCRG21) have been found. They can modulate (block) the vanilloid pain receptor TRPV1 and exhibit analgesic and anti-inflammatory effects in vivo [41,45-47].

Figure 4: (a) Multiple sequence alignment of Kunitz-type peptides: HCRG1 (C0HJU6) and HCRG2 (C0HJU7) [12], APHC1 (B2G331), APHC2 (C0HJF4) and APHC3 (C0HJF3) [38,46], InhVJ (P0DMJ5) [11], HCGS 1.10, HCGS 1.36, HCGS 1.19, HCGS 1.20 [10] from H. crispa, and BPTI (P00974) from Bos taurus. Identical and conservative amino acid residues are depicted on a dark and light gray background, respectively. Straight lines show three disulfide bridges. Alignment is done using Vector NTI. (b) The theoretical models of 3D structure of HCRG1. Model is presented in the form of ribbon diagram. In the core view, the disulfide bridges are visualized using Chimera.3D program.
According to a biosensor analysis, the native peptides, InhVJ, HCRG1, and HCRG2, represent highly specific inhibitors toward trypsin and α-chymotrypsin. They interact with the protease active center both canonical and alternative ways and do not inhibit other serine (thrombin, kallikrein, and plasmin), cysteine (papain) or aspartic (pepsin) proteases [11,12,43]. Three native peptides from the combinatorial library, APHC1, APHC2, APHC3 [38,46,48], and recombinant rHCRG21 [41], being weak trypsin inhibitors (with Ki in the range of magnitude 10-6-10-7 M), have been shown to modulate TRPV1 receptor. Wherein, APHC1 was the first peptide antagonist modulating TRPV1 detected in the venom of a sea anemone [38]. Recently, we have shown that rHCRG21 is the most effective peptide antagonist of TRPV1 [41]. “So, APHC1 inhibited ~32% of capsaicininduced currents at more than 200 nM [38], while APHC3 had a lower inhibitory effect (25%) even at higher concentrations more than 300 nM [45]. Unlike them, rHCRG21 has been shown to be a full blocker of TRPV1 (IC50 6,9 μM, maximal inhibition 95%) [41].
APHC1–APHC3, so called “analgesic peptides”, show in vivo an analgesic activity at doses of 0.01–0.1 mg/kg [38,39, 45] due to their inhibition of TRPV1. With that, these TRPV1 blockers demonstrate a significant analgesic effect without hyperthermia [39, 45].” Some other representatives of the combinatorial library, HCGS1.10, HCGS 1.19, HCGS 1.20, HCGS1.36 [10], have been shown to possess in vivo a pronounced analgesic effect in the model of thermal pain stimulation (“tail withdrawal”) [47]. In addition, when studying an anti-inflammatory activity on RAW 264.7 macrophages in the presence of lipopolysaccharide (LPS), rHCGS1.20 peptide shows an anti-inflammatory activity at the concentration of 10μM, it significantly reduces the level of NO by 25% [50]. The peptides HCRG1 and HCRG2 reduce the expression of inflammatory mediators, interleukin-1β (proIL-1β) in LPS-activated J774A.1 macrophage, as well as the secretion of interleukin-6 (IL-6) and the tumor necrosis factor TNF-α. However, these peptides do not reduce LPS-induced formation of NO [12]. Thus, the Kunitz-type peptides of H. crispa are promising analgesics and anti-inflammatory agents for scientific researches and pharmacology
Conclusion
The submitted minireview and the data presented in it do not contradict the long-established view that peptides from the sea anemone venom are highly selective, potent, and relatively safe compounds as potential therapeutic agents. This work was partially supported by research projects RFBR projects No 18-04-00631, No 18-38-00387, and “ERA”_RUS_ST2017-228.
References
- Madio B, King GF, Undheim EAB (2019) Sea Anemone Toxins: A Structural Overview. Mar Drugs 17(6): E325.
- Columbus Shenkar YY, Sachkova MY, Macrander J, Fridrich A, Modepalli V, et al. (2018) Dynamics of venom composition across a complex life cycle. eLife 7: e35014.
- de Lera Ruiz M, Kraus RL (2015) Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J Med Chem 58(18): 7093-7118.
- Jouiaei M, Yanagihara AA, Madio B, Nevalainen TJ, Alewood PF, et al. (2015) Ancient Venom Systems: A Review on Cnidaria Toxins. Toxins 7(6): 2251-2271.
- Liaoz Q, Fengz Y, Yang B, Lee S M Y (2019) Cnidarian peptide neurotoxins: a new source of various ion channel modulators or blockers against central nervous systems disease. Drug Discovery Today 24(1): 189-197.
- Zykova TA, Vinokurov LM, Kozlovskaya EP, Elyakov GB (1985) Amino-acid sequence of neurotoxin III from the sea anemone Radianthus macrodactylus. Russ J Bioorg Chem 11: 302-310.
- Kozlov SA, Osmakov DI, Andreev YA, Koshelev SG, Gladkikh IN, et al. (2012) Polypeptide toxin from sea anemone inhibiting proton-sensitive channel ASIC3. Russ J Bioorg Chem 38(6): 653-659.
- Monastyrnaya M, Leychenko E, Issaeva M, Likhatskaya G, Zelepuga E, et al. (2010) Actinoporins from the sea anemones, tropical Radianthus macrodactylus and northern Oulactis orientalis: Comparative analysis of structure-function relationships. Toxicon 56(8): 1299-1314.
- Isaeva MP, Chausova VE, Zelepuga EA, Guzev KV, Tabakmakher VM, et al. (2012) A new multigene superfamily of Kunitz-type protease inhibitors from sea anemone Heteractis crispa. Peptides 34(1): 88-97.
- Gladkikh I, Monastyrnaya M, Leychenko E, Zelepuga E, Chausova V, et al. (2012) Atypical reactive center Kunitz-type inhibitor from the sea anemone Heteractis crispa. Mar Drugs 10(7): 1545-1565.
- Gladkikh I, Monastyrnaya M, Zelepuga E, Sintsova O, Tabakmakher V, et al. (2015) New Kunitz-Type HCRG Polypeptides from the Sea Anemone Heteractis crispa. Mar Drugs 13(10): 6038-6063.
- Sintsova O, Gladkikh I, Chausova V, Monastyrnaya M, Anastyuk S, et al. (2018) Peptide fingerprinting of the sea anemone Heteractis magnifica mucus revealed neurotoxins, Kunitz-type proteinase inhibitors and a new β-defensin α-amylase inhibitor. J Proteomics 173: 12-21.
- Salceda E, Perez Castells J, Lopez Mendez B, Garateix A, Salazar H, et al. (2007) CgNa, a type I toxin from the giant Caribbean sea anemone Condylactis gigantea shows structural similarities to both type I and II toxins, as well as distinctive structural and functional properties. Biochem J 406(1): 67-76.
- Catteral WA, Beress L (1978) Sea anemone toxin and scorpion toxin share a common receptor site associated with the action potential sodium ionophore. J Biol Chem 253(20): 7393-7396.
- Zykova TA, Kozlovskaya EP (1989) Amino acid sequence of neurotoxin from the anemone Radianthus macrodactylus. Russ J Bioorg Chem 15(10): 1301-1306.
- Zykova TA, Kozlovskaya EP, Elyakov GB (1988) Amino acid sequence of neurotoxin II from the sea anemone Radianthus macrodactylus. Russ J Bioorg Chem 14(7): 878-882.
- Zykova TA, Kozlovskaya EP, Elyakov GB (1988) Amino acid sequence of neurotoxins IV and V from the sea anemone Radianthus macrodactylus. Russ J Bioorg Chem 14(11): 1489-1494.
- Kalina R, Gladkikh I, Peigneur S, Dmitrenok P, Zelepuga E, et al. (2019) Type II toxins from sea anemone Heteractis crispa with various effects on activation and inactivation of voltage-gated sodium channels. Toxicon 159: S18.
- Ortega Ramírez A, Vega R, Soto E (2017) Acid-sensing ion channels as potential therapeutic targets in neurodegeneration and neuroinflammation. Mediators Inflamm p. 1-18.
- Rash LD (2017) Acid-Sensing Ion Channel Pharmacology, Past, Present, and Future. Adv Pharmacol 79: 35-66.
- Kozlovskaya EP, Monastyrnaya MM, Gladkikh IN, Tabakmakher VM, Krivoshapko ON, et al. (2012) Polypeptide from sea anemone Heteractis crispa, having analgesic action.
- Kalina R, Gladkikh I, Dmitrenok P, Chernikov O, Koshelev S, et al. (2018) New APETx-like peptides from sea anemone Heteractis crispa modulate ASIC1a channels. Peptides 104: 41-49.
- Diochot S, Baron A, Rash LD, Deval E, Escoubas P, et al. (2004) A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO J 23(7): 516-1525.
- Gladkikh I, Kalina R, Koshelev S, Zelepuga E, Leychenko E, et al. (2019) Sea anemone Heteractis crispa produces a pool of peptides active on ASIC channels. Toxicon 159: S1-S32.
- Kristan K, Vierob G, Dalla Serra M, Maćek P, Anderluh G (2009) Molecular mechanism of pore formation by actinoporins. Toxicon 54(8): 1125-1134.
- Bakrać B, Anderluh G (2010) Molecular mechanism of sphingomyelin-specific membrane binding and pore formation by actinoporins. Adv Exp Med Biol 677: 106-115.
- Morante K, Bellomio A, Gil Cartón D, Redondo Morata L, Sot J, et al. (2016) Identification of a Membrane-bound Prepore Species Clarifies the Lytic Mechanism of Actinoporins. JBC 291(37): 19210-19219.
- Tejuca M, Anderluh G, Maćek P, Marcet R, Torres D, et al. (1999) Antiparasite activity of sea-anemone cytolysins on Giardia duodenalis and specific targeting with anti-Giardia antibodies. Int J Parasitol 29(3): 489-498.
- Laborde RJ, Sanchez Ferras O, Luzardo MC, Cruz Leal Y, Fernández A, et al. (2017) Novel adjuvant based on the pore-forming protein Sticholysin II encapsulated into liposomes effectively enhances the antigen-specific CTL-mediated immune response. J Immunol 198(7): 2772-2784.
- Mariottini GL, Pane L (2014) Cytotoxic and Cytolytic Cnidarian Venoms. A Review on Health Implications and Possible Therapeutic Applications. Toxins 6(1): 108-151.
- Leychenko E, Isaeva M, Tkacheva E, Zelepuga E, Malyrenko O, et al. (2018) Pore-forming toxins from sea anemone Heteractis crispa: diversity and pharmacological potential. Toxins 156: S9.
- García Linares S, Richmond R, García Mayoral MF, Bustamante N, Bruix M, et al. (2014) The sea anemone actinoporin (Arg-Gly-Asp) conserved motif is involved in maintaining the competent oligomerization state of these pore-forming toxins. FEBS J 281(5): 1465-1478.
- Fedorov S, Dyshlovoy S, Monastyrnaya M, Shubina L, Leychenko E, et al. (2010) The anticancer effects of actinoporin RTX-A from the sea anemone Heteractis crispa (=Radianthus macrodactylus). Toxicon 55(4): 811-817.
- Dai Y (2016) TRPs and pain. Semin Immunopathol 38(3): 277-291.
- Tabrizi MA, Baraldi PG, Baraldi S, Gessi S, Merighi S, et al. (2017) Medicinal Chemistry, Pharmacology, and Clinical Implications of TRPV1 Receptor Antagonists. Med Research Rev 37(4): 936-983.
- Gavva NR, Bannon AW, Hovland DN, Lehto SG, Klionsky L, et al. (2007) Repeated administration of vanilloid receptor TRPV1 antagonists attenuates hyperthermia elicited by TRPV1 blockade. J Pharmacol Exp Ther 323(1): 128-137.
- Andreev YA, Kozlov SA, Koshelev SG, Ivanova EA, Monastyrnaya MM, et al. (2008) Analgesic compound from sea anemone Heteractis crispa is the first polypeptide inhibitor of vanilloid receptor 1 (TRPV1). J Biol Chem 283(35): 23914-23921.
- Dyachenko IA, Andreev YA, Logashina Yu A, Murashev AN, Grishin EV (2015) Biological Activity of a Polypeptide Modulator of TRPV1 Receptor. Doklady Biological Science 465(1): 279-281.
- Philyppov IB, Paduraru ON, Andreev YA, Grishin EV, Shuba YM (2012) Modulation of TRPV1-dependent contractility of normal and diabetic bladder smooth muscle by analgesic toxins from sea anemone Heteractis crispa. Life Sci 91(19-20): 912-920.
- Monastyrnaya M, Peigneur S, Zelepuga E, Sintsova O, Gladkikh I, et al. (2016) Kunitz-type peptide HCRG21 from the sea anemone Heteractis crispa is a full antagonist of the TRPV1 receptor. Mar Drugs 14(12): E229.
- Zykova TA, Monastyrnaya MM, Apalikova OV, Shvets TV, Kozlovskaia EP (1998) Low-molecular cytolysins and trypsin inhibitors from sea anemone Radianthus macrodactylus. Isolation and partial characterization. Russ. J Bioorg Chem 24(7): 509-516.
- Sokotun IN, Leichenko EV, Vakorina TI, Eskov AA, Ilina AP, et al. (2007) A serine protease inhibitor from the anemone Radianthus macrodactylus: Isolation and physicochemical characteristics. Russ J Bioorg Chem 33(4): 448-455.
- Monastyrnaya M, Gladkikh I, Isaeva M, Zelepuga E, Sintsova O, et al. (2019) New Kunitz-type HCRG peptides of sea anemone Heteractis crispa. Mar Drugs 13(10): 6038-6063.
- Andreev YA, Kozlov SA, Korolkova YV, Dyachenko IA, Bondarenko DA, et al. (2013) Polypeptide modulators of TRPV1 produce analgesia without hyperthermia. Mar Drugs 11(12): 5100-5115.
- Kozlov SA, Andreev YA, Murashev AN, Skobtsov DI, Dyachenko IA, et al. (2009) New polypeptide components from the Heteractis crispa sea anemone with analgesic activity. Russ J Bioorg Chem 35(6): 789-798.
- Tabakmakher VM, Sintsova OV, Krivoshapko ON, Zelepuga EA, Monastyrnaya MM, et al. (2015) Analgesic effect of novel Kunitz-type polypeptides of the sea anemone Heteractis crispa. Dokl Biochem Biophys 461: 80-83.
- Nikolaev MV, Dorofeeva NA, Komarova MS, Korolkova YV, Andreev YA, et al. (2017) TRPV1 activation power can switch an action mode for its polypeptide ligands. PLoS ONE 12(5): e0177077.
- Sintsova ОV, Monastyrnaya ММ, Pislyagin EA, Menchinskaya ES, Leychenko ЕV, et al. (2015) Anti-inflammatory activity of the polypeptide of the sea Anemone, Heteractis crispa. Bioorg Khim 41(6): 657-663.
- Sintsova ОV, Pislyagin EA, Gladkikh IN, Monastyrnaya ММ, Menchinskaya ES, et al. (2017) Kunitz-type peptides of the sea anemone Heteractis crispa – potential anti-inflammatory compounds. Russ J Bioorganic Chem 43(1): 91-97.
