 Short Communication
Short CommunicationAbstract
The low glycemic index diet (LGID) was applied to seventeen patients who had previously been treated with the ketogenic diet (KD) but had demonstrated either partial (n=9) or no (n=8) improvement in seizure control. In 3 out of 8 patients in whom the KD failed, transition to the LGI diet led to reduction in seizure frequency by 30 to 70 %.In the group with an initial partial response to the KD, four out of 9 patients demonstrated a further reduction in seizure frequency while on the LGID, by 20, 20, 30 and 40% respectively. Two of the 9 patients maintained the seizure control previously achieved on the KD and the remaining 3 patients deteriorated/ presented an increase in number of seizures. During the LGID improvement in seizure control was combined with preservation of ketosis although markedly reduced in comparison to the KD and with reduced blood glucose below the level of 90 mg/dL. Deterioration of seizure control in a patient after an initial improvement during the LGID was combined with an elevation of blood glucose levels while ketone bodies remained unchangeable. In addition, patients who did not benefit from the LGID had difficulty in maintaining glucose levels under 90 mg/dL. Our data give evidence that the low glycemic index diet may have a different therapeutic mechanism from ketogenic diet.
Introduction
Over the last decades, the ketogenic diet (KD) has become a valuable weapon in the management of intractable seizures [1,2]. However not all of patients truly benefit from this therapy. In addition, the application of KD is limited by the fact that many patients cannot tolerate it while some others present with significant side effects [3-5]. Recently, many efforts have been made to improve its tolerability. For instance, a modified Atkins used by Kossof et al. is another good alternative of KD for treating patients with refractory epilepsy [6]. Other efforts are including the use of branched chain amino acids and creatine as adjunctive treatments to ketogenic diet [7,8]. Moreover, a low glycemic index diet (LGID) has been successfully used in patients with refractory epilepsy. The level of ketosis in the reported cases was less than that of the classical KD but still higher than that of conventional diets [9-12]. We present our experience of 17 patients who were initiated on the LGID. All these patients were previously following a traditional KD for a certain period of time but demonstrated partial or no improvement in seizure control. Our results are recorded and discussed in an effort to delineate the mechanism of action / the therapeutic effect of both diets.
Methods
Our study population consisted of the following groups of patients: Those with refractory epilepsy who were on ketogenic diet and their seizures were reduced by 50-80% (KD-responders, n= 9) and those in whom the KD failed to control their seizures (KD-non responders, n=8). Patients’ clinical and biochemical characteristics are presented for both groups in tables 1 and 2. A prerequisite for the inclusion of patients with partial improvement while on the KD was the stability on their condition defined as no change on the number of epileptic seizures for at least 3 months before taking part into the study. Non-response to ketogenic diet was defined as the failure of the KD to reduce seizure frequency when applied for at least 3 months. Moreover, patients included in the study did not have an overt inherited metabolic disorder, especially defects of b-acidosis. In all groups of patients, we gradually proceeded from ketogenic to low glycemic index diet while taking frequent measurements of their level of ketosis. As a principle was defined that in all patients under LGID plasma glucose levels should by maintained below ≤ 80 mg/dl.
Results

Table 1: Patients with non- response to KD
LGID=Low Glycemic Index Diet; KD=Ketogenic Diet; Glu=Glucose; b-OH-b=Beta-Hydroxybutyrate; AED=Antiepileptic Drugs.
In 14 patients the application of the LGID resulted in reduction of ketosis whereas in 3 other patients no change in the levels of β-hydroxybutyrate was observed. Two of them were belonging in the group that the ketogenic diet had complete failure while the other one in the group that ketogenic diet had partial success (Patients no 6 and 8 in the Table 1 and 8 in the Table 2). No one from these three patients was helped from the LGID. In 3 out of 8 patients in whom the KD completely failed, transition to the LGI diet managed to reduce seizure frequency by 30, 50 and 80% respectively (Table 1). The patient with the greatest reduction (80%) in seizure frequency, subsequently showed a relapse in the number of epileptic seizures the following 3 months which was combined with an increase in blood glucose levels from 70 to 100 mg/dl. When glucose levels decreased at 70 mg/dl, by reducing diet calories, the patient obtained the previous seizure control. (In the same time/during the study period) Despite fluctuations in seizure frequency and blood glucose levels during the study period, patient’s ketone body levels did not show any significant change. The other two patients remained in a stable improved condition for a time period longer than 6 months.
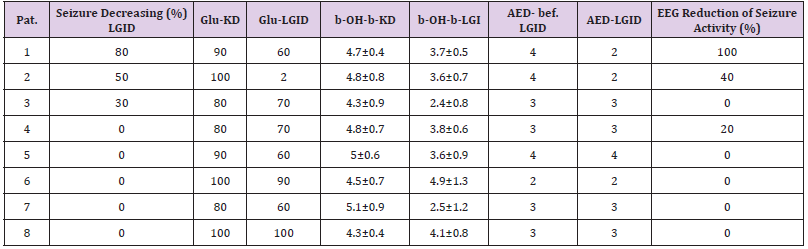
Table 2: Patients with partial- response to KD
LGID=Low Glycemic Index Diet; KD=Ketogenic Diet; Glu=Glucose; b-OH-b=Beta-Hydroxybutyrate; AED=Antiepileptic Drugs.
Four out of 9 patients with an initial partial response to the KD, demonstrated a further reduction in seizure frequency while on LGID, by 20, 20, 30 and 40% respectively (Table 2). The patient who presented with a 30% improvement, after a two-month period on the LGID (showed a relapse of episodes), returned to baseline seizure frequency associated with an increase in glucose levels from 80 to 100 mg/dl. To reduce blood glucose levels, we reduced the number of calories provided to the patient but without any success. In the same time, no significant change in the patient’s ketone body levels was observed. In another patient with an initial 20% reduction in seizure frequency during the first three months of the LGID, subsequently presented with an increase in the number of epileptic episodes along with an elevation of blood glucose from 60 to 90 mg/dl, although no change in his diet had occurred. Reduction in the number of calories provided was combined with a concomitant reduction in the number of seizures and fall of glucose levels. During the study period no adverse effects were recorded in our patients due to transition from the KD to the LGID (initiated on the LGID).
Discussion
Our first observation was that in 14 patients, application of the LGID resulted in reduction of ketosis whereas in 3 other patients no change in the levels of β-hydroxybutyrate was observed. One of them belonged in the team that the ketogenic diet had partial success while the other two in the team that ketogenic diet had complete failure. In the absence of an overt cause for this difference, we speculated that unchangeable ketone body levels were due to a potential unknown disturbance in glucose metabolism. This is further supported by the fact that two out of 3 patients had practically no change in glucose levels. Also, to all of these three patients, the application of LGID had totally failed to help them. May be, although the number of these patients is too small, we may hypothesize that difficulties in lowering the glucose levels is a bad predictive factor for the application of the low-glycemic index diet.
There are many data, experimental and in human subjects, where disturbed glucose metabolism has been described in refractory epilepsy [13-15]. Abnormal oral glucose tolerance test is frequently seen in patients with intractable epilepsy [16] and this might be the case with our patients especially those two in whom neither ketone bodies nor blood glucose could be decreased. As mentioned above, among the group of KD-non responders, there were three patients who benefited by the transition to the LGID. In all three, we observed a concomitant decrease in both β-hydroxybutyrate and glucose levels. It should be strained here/ mentioned that ketone bodies although markedly decreased, did not reach normalization (<1,5mg/dl). Thus, seizure control does not seem to correlate with ketonemia as much as with reduced blood glucose. This speculation/ observation is in accordance with the fact that deterioration of seizure control in a patient was combined with an elevation of blood glucose levels while ketone bodies remained unchangeable. When blood glucose returned to desired reduced levels, a satisfactory reduction in seizure frequency was achieved. Another parameter that should be reported/ mentioned /considered is that in order to reduce glucose levels, the patient was put on a more calorie-restricted diet.
As a result, we are not sure whether the factor contributing to the patient’s improvement is the decrease of Glucose levels or the restriction on calories or the combination of both factors. We have experimental data where the reduction of the ingesting calories has beneficial effects on the number of crises [17]. In the group where the KD had some success, four patients presented further improvement. In the patients where we initially observed a decrease of ketosis, we had a reduction of crises. Here we must notice, that although the ketosis reduced, the patients remained ketotic. The same patients showed a reduction of glucose with their glucose levels stably remaining under 80 mg/dl. We wondered about the cause of improvement in these four patients. Was it the reduction of ketosis or the reduction of glucose levels or both combined? Taking into consideration the number of patients we have; it is difficult to find an answer to this question. Here, it is interesting to observe that all four patients presented an increase of convulsions after a stable period of three to five months.
However, in the same group some of the patients relapsed without displaying a reduction of ketosis. What we certainly noticed was an elevation of glucose levels to >100 mg/dl. Subsequent some of them demonstrated a new improvement not combined with a fluctuation of ketosis but with a reduction of glucose levels. Reciprocally was it impossible to attain a further reduction of the glucose levels with patients who did not display improvement, even by reducing the number of calories. The first question to be answered is why we had an elevation of glucose levels although nothing in the diet changed. Was the elevation of glucose levels a result of the convulsions or was it something that happened before the appearance of convulsions? The last question is one that considering the data we have is difficult to be answered. From experimental models we know that the acute appearance of convulsions increases the glucose metabolism. But we are also aware of the increased glucose metabolism due to chronic unregulated epilepsy. Therefore, it is indicated, that both the acute and the chronic resisting epilepsy are accompanied by the hypermetabolism of glucose [18-20]. This theory is supported by the fact that 2-deoxy-D-glucose (2DG), a substance that inhibits glycolysis has antiepileptic effects [21-24].
Conclusion
In conclusion, we believe that although the diet with a low glycemic index and the KD have some similarities, they display different mechanisms contributing to their outcome. This is indicated by the fact that people who negatively responded to the KD, displayed improvement with the diet with a low glycemic index, whereas patients who positively responded to the KD deteriorated after the application of the LGID. As a result, it is demonstrated that some people’s element contributing to their success is the ketosis while others appears to be a decreased glucose level. It is even possible, that other patients’ therapeutic success is related to both factors while others adequate either a low degree of ketosis or the restriction on the number of calories. What factor achieved higher therapeutic success must do also with the individual characteristics in the metabolism of each person. Therefore, there must be made more intensive research about the individual characteristics of every single person’s metabolism. This will help us to make the existing diets more efficient, to develop new diets, or to allocate the appropriate diet to the appropriate person.
Conflicts of interest
The authors have no conflict of interest to declare
References
- De Campo DM, Kossoff EH (2019) Ketogenic dietary therapies for epilepsy and beyond. Curr Opin Clin Nutr Metab Care 22(4): 264-268.
- Winesett SP, Bessone SK, Kossoff EH (2015) The ketogenic diet in pharmacoresistant childhood epilepsy. Expert Rev Neurother 15(6): 621-628.
- Lin A, Turner Z, Doerrer SC, Stanfield A, Kossoff EH (2017) Complications During Ketogenic Diet Initiation: Prevalence, Treatment, and Influence on Seizure Outcomes. Pediatr Neurol 68: 35-39.
- Kang HC, Chung DE, Kim DW, Kim HD (2004) Early- and late-onset complications of the ketogenic diet for intractable epilepsy. Epilepsia 45(9): 1116-1123.
- Ballaban-Gil K, Callahan C, O'Dell C, Pappo M, Moshé S, et al. (1998) Complications of the ketogenic diet. Epilepsia 39(7): 744-748.
- Kossoff EH, Cervenka MC, Henry BJ, Haney CA, Turner Z (2013) A decade of the modified Atkins diet (2003-2013): Results, insights, and future directions. Epilepsy Behav 29(3): 437-442.
- Evangeliou A, Spilioti M, Doulioglou V, Kalaidopoulou P, Ilias A, et al. (2009) Branched chain amino acids as adjunctive therapy to ketogenic diet in epilepsy: pilot study and hypothesis. J Child Neurol 24(10): 1268-12
- Kalamitsou S, Masino S, Pavlou E, Gogou M, Katsanika I, et al. (2019) Effect of creatine supplementation on seizure control in children under ketogenic diet: A pilot study. Integr Mol Med 6: 1-6.
- Pfeifer HH, Thiele EA (2005) Low-glycemic-index treatment: A liberalized ketogenic diet for treatment of intractable epilepsy. Neurology 65(11): 1810-1812.
- Kim SH, Kang HC, Lee EJ, Lee JS, Kim HD(2017) Low glycemic index treatment in patients with drug-resistant epilepsy. Brain Dev 39(8): 687-692.
- Grocott OR, Herrington KS, Pfeifer HH, Thiele EA, Thibert RL (2017) Low glycemic index treatment for seizure control in Angelman syndrome: A case series from the center for Dietary Therapy of Epilepsy at the Massachusetts General Hospital. Epilepsy Behav 68: 45-50.
- Muzykewicz DA, Lyczkowski DA, Memon N, Conant KD, Pfeifer HH, et al. (2009) Efficacy, safety, and tolerability of the low glycemic index treatment in pediatric epilepsy. Epilepsia 50(5): 1118-1126.
- Mantis JG, Centeno NA, Todorova MT, McGowan R, Seyfried TN (2004) Management of multifactorial idiopathic epilepsy in EL mice with caloric restriction and the ketogenic diet: role of glucose and ketone bodies. Nutr Metab (Lond) 1(1):
- Theodore WH, Kelley K, Toczek MT, Gaillard WD (2004) Epilepsy duration, febrile seizures, and cerebral glucose metabolism Epilepsia 45(3): 276-279.
- Tang Y, Liow JS, Zhang Z, Li J, Long T, et al. (2019) The Evaluation of Dynamic FDG-PET for Detecting Epileptic Foci and Analyzing Reduced Glucose Phosphorylation in Refractory Epilepsy. Front Neurosci 12: 993.
- Varlamis S, Vavatsi N, Pavlou E, Kotsis V, Spilioti M, et al. (2013) Evaluation of Oral Glucose Tolerance Test in Children with Epilepsy. J Child Neurol 28(11): 1437-1442.
- Raffo E, François J, Ferrandon A, Koning E, Nehlig A (2008) Calorie-restricted ketogenic diet increases thresholds to all patterns of pentylenetetrazol-induced seizures: critical importance of electroclinical assessment. Epilepsia 49(2): 320-328.
- Mirrione MM, Schiffer WK, Siddiq M, Dewey SL, Tsirka SE (2006) PET imaging of glucose metabolism in a mouse model of temporal lobe epilepsy. Synapse 59(2): 119-121.
- Cornford EM, Nguyen EV, Landaw EM (2000) Acute upregulation of blood-brain barrier glucose transporter activity in seizures. Am J Physiol Heart Circ Physiol 279(3): H1346-1354.
- Wasterlain CG, Fujikawa DG, Penix L, Sankar R (1993) Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia 34(Suppl 1): S37-53.
- Gasior M, Yankura J, Hartman AL, French A, Rogawski MA (2010) Anticonvulsant and proconvulsant actions of 2-deoxy-D-glucose. Epilepsia 51(8): 1385-1394.
- Stafstrom CE, Ockuly JC, Murphree L, Valley MT, Roopra A, et al. (2009) Anticonvulsant and antiepileptic actions of 2-deoxy-D-glucose in epilepsy models. Ann Neurol 65(4): 435-447.
- Stafstrom CE, Roopra A, Sutula TP (2009) Seizure suppression via glycolysis inhibition with 2-deoxy-D-glucose (2DG). Epilepsia 49 (Suppl 8): 97-100.
- Long Y, Zhuang K, Ji Z, Han Y, Fei Y, et al. (2019)2-Deoxy-D-Glucose Exhibits Anti-seizure Effects by Mediating the Netrin-G1-KATP Signaling Pathway in Epilepsy. Neurochem Res 44(4): 994-1004.
