 Case Report
Case ReportAbstract
Polyarteritis nodosa (PAN); is an uncommon vasculitis involving different organ systems with an estimated prevalence of 31 cases per million. Cutaneous polyarteritis nodosa is again a rare variant of PAN accounting for approximately 4 per cent of PAN cases. Here we present a case of a 32 years old male with the appearance of multiple discolored nodules, initially misdiagnosed as erythema nodosum, without any constitutional features and systemic involvement. His diagnosis was confirmed with a skin biopsy and his symptoms responded to high dose steroids and cyclophosphamide. The purpose is not only reporting a rare case but also to enhance the clinical knowledge of the non-dermatologists’ physicians regarding the unusually benign course and more favorable prognosis of this rare variant of a destructive vasculitis.
Keywords: Cutaneous Polyarteritis Nodosa: A Rare Variant of a Destructive Vasculitis
Introduction
Polyarteritis nodosa (PAN) is classically characterized by segmentary leukocytoclastic vasculitis affecting the small- and medium-sized arteries. In 1931, Lindberg described a cutaneous variant of PAN without visceral involvement and with a more favorable prognosis.
Case Presentation
A 32 years old male presented with a 1-week history of a lump in the right axilla along with on/off chest pain and shortness of breath. According to him, the lump was painless and has been decreasing in size since he first noticed it. The patient had a significant past history of multiple recurrences of erythematous nodules predominantly on the left leg, which was previously diagnosed as erythema nodosum. Travel and sexual history were noncontributory. On examination, he was vitally stable with a blood pressure of 120/80mm of Hg and a regular heart rate of 95/min. Rest of his systemic examination was insignificant for any findings except for a small, rubbery and non-tender lump in the right axilla. The investigations including CBC, TFTs, and urinalysis were normal with no monoclonal proteins in the urine. The erythrocyte sedimentation rate was borderline high at 17 mm/at the end of the 1st hour. Hepatitis B and C testing were negative. Immunological investigations including c-ANCA and p-ANCA were negative with normal complement levels. A biopsy of from the skin of the left thigh was taken and histopathological assessment showed lobular panniculitis, granulomatous inflammation and vasculopathy supporting the diagnosis of polyarteritis nodosa (Figures 1 & 2). He was started on high dose steroids i.e. oral prednisolone 1mg/kg along with 3 cycles of cyclophosphamide every month. The steroid was then tapered off slowly. His lesion disappeared and he was improved clinically.
Discussion
Cutaneous Polyarteritis Nodosa is a variant of polyarteritis nodosa which mainly affects the skin, however constitutional symptoms like fever, muscle and joint involvement may also occur [1-3]. Major organs are usually spared in this disorder. Typical cutaneous lesions of PAN i.e. nodules, livid, ulceration and purpura are present. Histologically, the arteries of deep dermis or hypodermis shows leukocytoclastic vasculitis with or without associated fibri noid necrosis and visceral involvement is absent at the time of diagnosis [1]. Like other inflammatory disorders, ESR is usually the only laboratory parameter altered. The presence of leukocytosis and eosinophilia, persistent proteinuria, high blood pressure, and diffuse neuromuscular involvement makes the systemic PAN distinct from the cutaneous variant and all these factors results in the poor short term of prognosis of systemic PAN [4]. According to the diagnostic criteria of the American College of Rheumatology, a disease with one cutaneous and at least one extra-cutaneous symptom with appropriate histopathological findings is referred to as PAN [5].
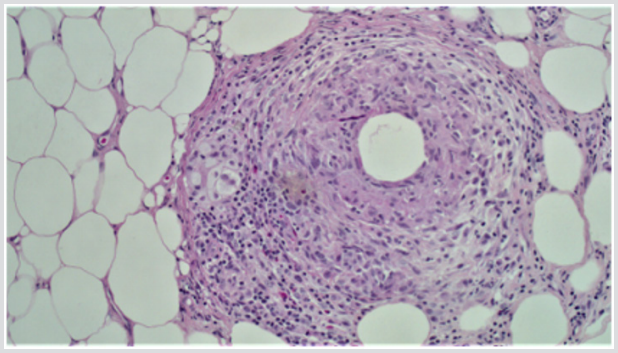
Figure 1: Skin biopsy showing lobular panniculitis, granulomatous inflammation and vasculopathy.
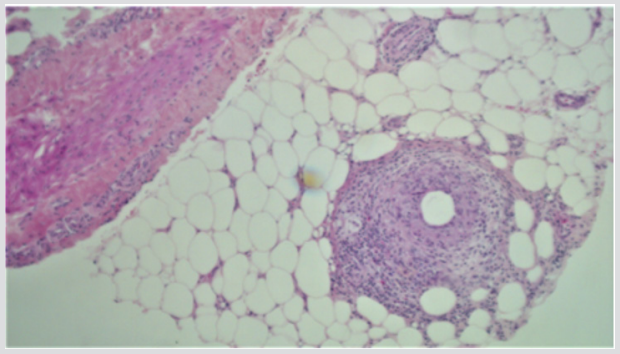
Figure 2: Skin biopsy showing granulomatous inflammation and vasculopathy.
In 25-30% patients of systemic PAN, cutaneous involvement can be seen. The microscopic findings of cutaneous PAN can be divided into four stages:
a. Degenerative stage [6]
b. Acute inflammatory stage [7]
c. Granulation tissue stage and
d. Healed end-stage [8,9].
Unlike the systemic PAN, aggressive treatment is not required in Cutaneous PAN to achieve the remission phase and usually a conservative therapeutic plan is followed. Although there are no double-blind prospective placebo-controlled trials for therapy, treatment could be initiated with low doses of sulphapyridine, NSAIDs or salicylate that may induce remission or relieve the symptoms or signs of the disease [10]. Antibiotic treatment may be needed in patients with secondary bacterial infections [11]. If there is no response, low or moderate doses of corticosteroids or weekly methotrexate have been used with good responses [12].
Conclusion
In conclusion, c-PAN is a benign, rare variant of systemic PAN restricted usually to the medium-sized arteries of the subcutis, lacking neuromuscular and constitutional symptoms in most of the cases and unlikely to progress to systemic PAN. It is important that the patient is followed up closely in order to notice any unusual case in which the patient might progress to systemic PAN.
References
- Maillard H, Szczesniak S, Martin L (1999) Périartérite noueuse cutané Particularités diagnostiques et thérapeutiques de 9 cas. Ann Dermatol Venereol 126: 125-129.
- Daoud MS, Hutton KP, Gibson LE (1997) Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol 136(5): 706-713.
- Lindberg K (1931) Ein Beitrag zur Kenntnis der Periarteritis nodosa. Acta Med Scand 76: 183-225.
- Machet L, Szczesniak S, Callens A, de Muret A, Lorette G (1993) La périartérite noueuse cutanée: étude rétrospective de 9 cas. La Revue de Médecine Interne 14(6): 403.
- A Bauzá, A España M Idoate (2002) Cutaneous polyarteritis nodosa. British Journal of Dermatology 146(4): 694-699.
- Nakamura T, Kanazawa N, Ikeda T, Yamamoto Y, Nakabayashi K, et al. (2009) Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res 301(1): 117-121.
- Borrie P (1972) Cutaneous polyarteritis nodosa. British Journal of Dermatology 87(2): 87-95.
- Diaz Perez J (1974) Cutaneous Periarteritis Nodosa. Archives of Dermatology 110(3): 407-414.
- Fauci AS (1978) The spectrum of vasculitis. Ann Intern Med 89(5 Pt 1): 660-676.
- Diaz Perez JL, Winkelmann RK (1980) Cutaneous periarteritis nodosa: a study of 33 cases. In: Vasculitis (Wolff K, Winkelmann RK (eds.). London: Lloyd-Luke pp. 273-284.
- Daoud MS, Hutton KP, Gibson LE (1997) Cutaneous periarteritis nodosa: a clinicopathological study of 79 cases. Br J Dermatol 136(5): 706-713.
- Jorizzo JL, Sherertz EF, Zanolli MD, Zanolli MD, Sherertz EF (1991) Low dose weekly methotrexate forunusual neutrophilic vascular reactions: Cutaneous polyarteritis nodosum and Behcet’s disease. J Am Acad Dermatol 24(6 Pt 1): 973-978.
