 Review Article
Review ArticleAbstract
The molecular diagnosis of blood infections is challenging because pathogens exist in the bloodstream in very low concentrations. Consequently, such sparse target populations require DNA extraction and purification from large volume biofluids. This work utilizes the advances in microfluidic technologies to demonstrate the nucleic acid purification - qPCR sequence detection workflow for this application. Immiscible Phase Filtration (IPF) for nucleic acid purification and Electrowetting-on Dielectric (EWD) droplet actuation are combined on a hybrid microfluidic device that translates from large volume sample-to-small-volume analysis. After IPF reduces the sample volume from a milliliter-sized lysate to a microliter-sized eluent, EWD can be used to automatically prepare the PCR mixture. This step begins with transporting droplets of the PCR reagents to mix with the eluent droplets. Suitable assays, microfluidic stages, and auxiliary systems are described. The extent of purification obtained per IPF wash, and hence the number of washes needed for uninhibited qPCR, are determined via onchip UV absorbance. The performance of on-chip qPCR, particularly the copy number to threshold cycle correlation, is characterized. Lastly, the above developments accumulate to an experiment that includes the following on-chip steps: DNA purification by IPF, PCR mixture preparation via EWD, and target quantification using qPCR - thereby demonstrating the core procedures in the proposed approach.
Introduction
The requirement to detect trace, quantifiable amounts of DNA in large-volume biomedical samples requires advanced DNA preparation techniques using modern technologies to facilitate DNA isolation, purification and analysis by Quantitative PCR (qPCR). Thus, a key step in DNA analysis is high efficiency Nucleic Acid (NA) purification. Conventional laboratory-based magnetic bead purification involves repetitive pipetting and centrifugation. For example, depending on the starting sample, as many as seven washes may be necessary for removing the contaminants entrapped in the bead pellet, adsorbed on the walls of reaction tubes, or remaining in the solution after aspirating the supernatant [1]. As a result, without access to costly robots this process is labor intensive.
Early Work
The suitability of microfluidic technology in performing standard DNA extraction and purification protocols with integration into downstream PCR amplification, separation and sample preparation has been investigated by numerous workers. Morales and Zahn [2] reviewed microfluidic methods for NA purification and demonstrated NA purification using droplet-based flow in a serpentine device. However, their device was not integrated with any other steps in a DNA analysis protocol. Several other chip-based structures have been proposed that use silica-based separations and magnetic beads for extraction within systems that combine cell lysis, DNA extraction, and PCR [3-6]. However, the DNA must be eluted prior to PCR, which has been problematic due to the presence of PCR inhibitors in the samples. In addition, since most reported devices are limited to only a few reaction chambers, multiplex detection of multiple pathogen targets is difficult [3- 8]. Devices comprising 12 reaction chambers for multiplexed detection have been described, but these devices did not integrate cell lysis and DNA extraction [9]. A recent review of the use of digital microfluidics for PCR is presented in Ref. [10].
A droplet-based CMOS chip has been described with integrated heaters and sensing for performing qPCR [11]. The chip had onboard reservoirs for dispensing primers, PCR reagents, and pre-processed DNA. However, no attempt was made to extract and purify DNA on chip. A key issue in integrated devices is DNA purification prior to qPCR. Kim and Gayle [12] demonstrated extraction with nano-filtration of lysed whole blood samples with an integrated Aluminum-Oxide Membrane (AOM). An elution buffer was then flowed through the AOM and DNA was collected for PCR. However, extracted DNA efficiency was only 5% of the anticipated DNA in the blood sample, indicating a lack of full recovery from the membrane. As a result, qPCR could not be demonstrated with this system. Oblath et al. [13] also described a microfluidic chip that integrated DNA extraction using an AOM, followed by amplification and detection. Extraction of DNA was performed by creating a pressure gradient across a nano-porous Aluminum- Oxide Membrane (AOM) located at the bases of 10μL reaction wells. Sample fluid flow through the AOM allowed for DNA extraction in the presence of an applied vacuum without the need for elution, but with user intervention. PCR reagents were then added to the wells and PCR was performed with the PCR solutions located just above the AOM. Whereas the AOM did not inhibit PCR results, there was no indication of extracted DNA efficiency. Oblath et al. did not demonstrate Quantitative PCR (qPCR), and they only point out that quantitative real-time data for diagnostics may be possible using their approach.
Other methods of DNA purification suitable for qPCR in a microfluidic implementation have been proposed. Berry et al. [14] demonstrated NA sample purification in a micro channel using an immiscible phase to filter contaminants in a single step. The processes used magnetic beads bound with NA, which were magnetically actuated across the immiscible barrier of a separate phase. Purification was achieved in reduced time by a mechanism that relied on surface-tension at a small scale. A non-microfluidic demonstration of Immiscible Phase Filtration (IPF) for NA purification was made by Sur et al. [1] Regarding the immiscible phase, compounds such as air [15], silicone oil [16,17], mineral oil [18], and liquid wax [1] have been used.
Bienvenue et al. discussed an integrated microfluidic device for DNA purification and PCR [19]. The device performed purification of DNA from a complex biological sample (whole blood) and timed DNA elution from a silica solid phase for downstream PCR amplification by placement of the micro device into a conventional thermocycler. Thus, there was no on-chip qPCR capability. In other work, Mahalanabis et al. [20] demonstrated an integrated microfluidic device that combined chip-based sample preparation and isothermal amplification. DNA extraction was performed using micro-Solid-Phase Extraction (μSPE) in a bead column fabricated in a thermo-plastic device. No mention was made of handling sparse samples in large volumes or sample-to-answer throughput using qPCR.
The use of immiscible phase filtration in NA purification and qPCR also has been claimed in a droplet platform in which droplet actuation occurred by Electrowetting on Dielectric (EWD) [21]. Magnetic-capture beads in a droplet were constrained using an external magnet, and the beads as well as excess liquid in the droplet surrounding the beads could be “snapped off” by transporting the droplet away from the beads. Quantitative PCR was demonstrated in [21] after eluting DNA from the beads into an on-chip reservoir. However, such droplet-only-based systems are constrained to be used with small multiples of the nanoliter volumes typical for EWD chips [22].
In light of the above reports, the present work demonstrates a hybrid microfluidic device aimed at quantitative diagnostics to detect sparse pathogen targets in large volumes of biofluids. Importantly, rather than electing to optimize a particular diagnostic process in sparse pathogen analysis, microfluidic technologies should be applied to expedite and automate the entire workflow for efficient throughput. Towards this end, a compatible set of microfluidic technologies is presented that are combined to expedite and automate NA purification and qPCR sequence detection. The present work builds on concepts initially presented by Hsu et al in 2012 [23]. Additional details are presented in [24]. Importantly, although a full-fledged diagnostic device is beyond the scope of this work, the employed approach nonetheless should be scalable to meet the demands of, for example, blood infection diagnosis.
Microfluidic Device Requirements
A primary objective in the present work was to fabricate a device that was capable of detecting multiple types of low-concentration pathogens from a large-volume raw sample. The large sample size favors the use of IPF, since the method can process a millilitersized sample and output the resulting purified NA in a way that is compatible with small volume, second phase qPCR steps.
As discussed above, Immiscible Phase Filtration (IPF) is a variant of the conventional bead-based assay [1,14,18]. A general schematic view of how IPF was incorporated into our hybrid microfluidic system is shown in Figure 1. Beads bound with NA are first snapped out of the lysate to an immiscible phase. Next, the beads are dispersed in a wash buffer. Here, the contaminants originally entrapped in the bead pellet are diluted away. Simultaneously, proteins adsorbed on the beads are denatured by chaotropic salts in the wash buffer [23].
After washing, the beads are transported from the wash buffer through an immiscible phase to an elution buffer. In this process, it is energetically unfavorable for contaminants, such as the chaotropic salts, to enter the immiscible phase, since IPF functions as a series of magneto-capillary valves [25,26]. Carryover volume has been shown to be linearly proportional to bead load, i.e., total bead weight [1]. In consequence, to reduce the bead load (thus, the carryover) without sacrificing the surface area for NA binding, smaller magnetic beads are advantageous. Still, since the magnetic force acting on a magnetic bead is proportional to its volume, it is relatively difficult for smaller beads to overcome the interfacial tension and achieve bead snapping.
The interface between immiscible phase filtration and the elution step is an Elution Buffer (EB) reservoir, as illustrated in Figure 1. The EB reservoir is the last section of the purification stage (stores the elution buffer) that doubles as the first section of the PCR preparation stage (stores the eluent and then dispenses the eluent droplets.) Specifically, during elution nucleic acid-bound magnetic silica beads are transported to the EB reservoir. Then, due to the low-salt condition in the reservoir, the NA is released from the beads to the EB. After a brief incubation period, the beads are transported away from the EB reservoir to prevent the beads from lowering the PCR amplification efficiency. Afterwards, eluent droplets can be dispensed from the EB reservoir for the subsequent PCR mixture preparation. The EB reservoir is implemented on a Digital Microfluidic (DM) platform where fluid flow is performed by droplet actuation using Electrowetting-On-Dielectric (EWD) [21]. DM control is well-suited to prepare the PCR mixtures and to perform qPCR, since EWD actuation is inherently automated, which minimizes hands-on time, eliminates operator error, and improves reproducibility [21,27].
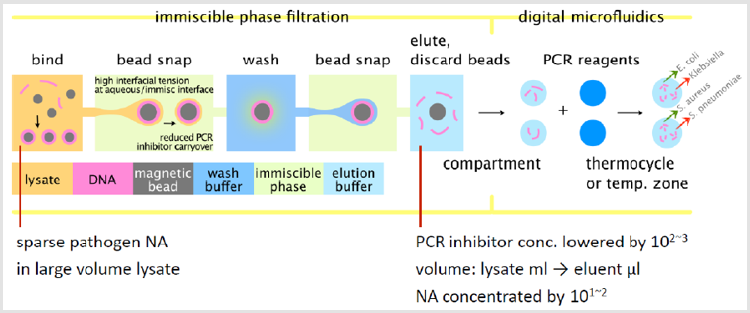
Figure 1: Schematic of an IPF device, top-down view. NA from samples in milliliter volumes is purified from the lysate as the magnetic beads are transported to the right through the device, washed to remove PCR inhibitors, and DNA is eluted for no-chip qPCR and multiplex detection.
Experimental
Two generations of microfluidic devices were fabricated and tested. The first-generation device is described in [23,24]. A sketch of the layout of the second-generation device is shown in Figure 2. A photo of the experimental device is shown in Figure 3. The experimental device consisted of a Secure Seal double-sided adhesive film (620001, Grace Bio-Labs) sandwiched by two Cytopcoated acrylic plates. The top acrylic plate was CNC-milled with pipette ports for loading reagents into reservoirs, whereas the bottom acrylic plate was featureless. The Secure Seal was laser -patterned to achieve the desired pattern to perform purification by IPF and placed on the bottom acrylic plate. The immiscible phase chosen was air. The output of the IPF section was the elution buffer that was assessable through an air/silicone oil interface. After loading the EB, droplets were dispensed in the 5cSt silicone oil medium.
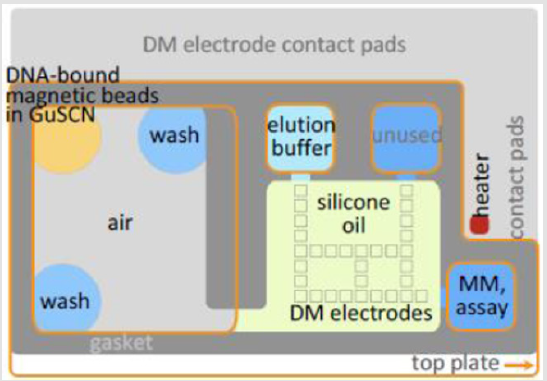
Figure 2: A sketch showing the layout of Generation II fluidic device.

Figure 3: Photo of hybrid microfluidic device.
The EWD section was designed to generate 46nL droplets on 700μm electrodes and a channel height of 120μm. A crosssection of the EWD droplet actuator is shown below in Figure 4. The conductive ground plane of the top acrylic plate was ITO/ PEDOT: PSS on acrylic in the thicknesses shown in Figure 4. This combination of films mainly was necessary to accommodate the CTE mismatch between ITO and acrylic due to the high temperatures needed during PCR (95oC). Subsequently, both top and bottom plates were spin-coated with Cytop to form hydrophobic surfaces. The hybrid device was then assembled by pressing the top plate against the Secure Seal bottom plate. Finally, assembled devices were baked overnight at 80oC to evaporate Cytop solvent and to enhance Secure Seal adhesion.
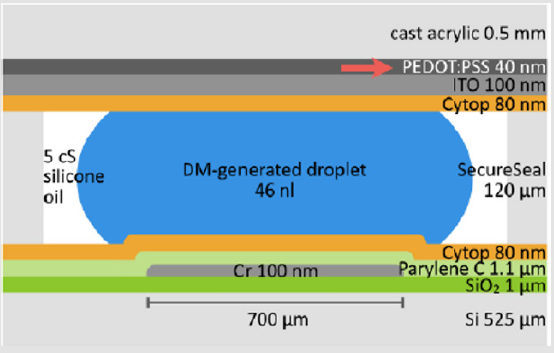
Figure 4: Cross-section of the EWD droplet actuator in the qPCR device.
The overall strategy in performing DNA purification by IPF and qPCR on a EWD device is described above. The demonstration of on-chip NA purification and quantification necessitated three types of reagents: qPCR assay, PCR master mix, and the purification kit. On-chip purification was performed on the experimental device in the following steps [23]:
a) Preparation of gDNA-Bead Wash Buffer Suspension
In the first step, HeLa gDNA was bound to magnetic silica beads. First, 60μl of 1ng/μl gDNA (about 1.3105x copies of Yb8) and 50μl of Magnetic Extraction Reagent (miniMAG) were added to 1 ml Lysis Buffer (LB). This step was performed off-chip, since on-chip milliliter volume cell lysing was not yet available. After incubating and centrifuging, the LB Buffer supernatant was aspirated. The DNA-bound beads were then resuspended with Wash Buffer1 (WB1), i.e., 5M GuSCN. Lastly, the DNA-bound beads were resuspended with 400μl WB1 (i.e., 5M GuSCN.) To ensure the amount of Yb8 is consistent across the replicates, the suspension was vortexed before 4μl (about 1:3 _ 103x copies) was transferred to the device. Assuming all Yb8 is retained, a reaction mixture droplet would possess 30x copies.
b) Reagent Loading
In the second step the reagents were loaded into the microfluidic device shown in Figure 2. Prior to an experiment, the DNA-bead reservoir and wash-buffer3 reservoirs (WB3 -two locations) were loaded on the purification stage, and the EB reservoir was filled in the PCR preparation stage.
c) Elution
The last step was elution. After the two washes, the beads were transferred from the second WB3 reservoir through an air/silicone oil interface to the EB reservoir in the PCR preparation stage. To release the DNA from the beads to the EB reservoir, the beads were incubated in the EB at room temperature.
d) qPCR Assay
The qPCR assay was comprised of two primers and a fluorophore-labelled probe. The dye on the probe was selected to be HEX, Quantitative PCR was performed in the EWD section of the experimental device in the following steps [23]. On-chip qPCR was performed on 46nl droplets of eluent by cycling the temperature of an on-chip heater. Droplets of eluent were dispensed on the EWD device and mixed with a dispensed droplet of qPCR assay mix. The combined 2x droplet was transported to a location adjacent to the heater. (See Figure 2) because its absorption peak is nearly coincident with the 532 nm output of low-cost laser diodes. Also, it is one of the stock dyes available from the major oligomer vendors. Since off-the-shelf qPCR assays are typically only available with FAM (494/518 nm), in order to replace FAM with HEX it was necessary to involve custom qPCR assays. Two assays designed to detect multicopy/genome targets were identified to be useful:(1) Alu Yb8 detects an 1852 copy/genome human-specific 176 bp sequence [28,29].
(2) EC23S857 detects a 6 copy/genome E. coli-specific 88 bp target [30].
After the Yb8 assay was selected and synthesized (by Integrated DNA Technologies, Inc.), qPCR was performed to validate the design and the synthesis. Specifically, the assay was employed to quantify the Yb8 in the NA extracted from HeLa cells using the miniMAG purification kit. The Fast-Advanced master mix was used to prepare the reaction mixtures, and the amplification was run on an ABI 7900HT benchtop qPCR instrument. Successful amplification was achieved, which suggests that the assay was correctly designed and synthesized. Further, from a calibration curve the amplification efficiency was calculated to be 81%. Lastly, although the FAM -labelled probe was used in this evaluation in order to maintain the compatibility with the ABI 7900HT, the HEX-labelled probe was used in all on-chip qPCR tests.
Two master mixes were evaluated: QuantiFast qPCR kit [30] and Fast Advanced Master Mix [32]. Although most experiments herein were conducted using Fast Advanced, in practice the main differences between Quanti Fast and Fast Advanced essentially was the flexibility of the reference dye and the ability to prevent cross contamination. A 40μl stock was prepared from 20μl Fast Advanced master mix, 2μl Yb8 assay, and 18μl nuclease -free water. The water was added to keep the final master mix and assay concentrations the same.
The purification assay was the ensemble of reagents used to perform cell lysis and the subsequent purification of NA from the lysate. Here, the employed purification assay was bio Meraux’s Nuclei Sens line of Lysis Buffer and Magnetic Extraction Reagents [33]. Together, they were referred herein as the miniMAG assay [34]. After comparing six bead-based purification kits, miniMAG was selected because it was designed to lyse and purify the chaotrope GuSCN and the surfactant Triton X-100, but without the help of Proteinase K [35]. In contrast to the kits that mandate Proteinase K treatment at 56oC, miniMAG makes it possible to initiate the future testing of on-chip lysis without the need to add an extra heater. The temperature control system was a LabVIEW program that relied on the feedback from a temperature sensor to conduct two functions. First, the system regulates the on-chip heater to carry out thermocycling. Second, when cooled to a pre-set temperature, it will trigger the fluorescence sensing system to perform spectrum acquisition. The LabVIEW program was composed of two finite state machines: thermocycler FSM and fluorometer FSM. In particular, the thermocycler FSM controls the on-chip heater using the PID algorithm.
The on-chip heater and the EWD electrodes in Figure 2 were located in the same metal layer using 100nm e-beam evaporated Cr. It was found that placing the heater near the EWD electrodes and hence close to the aqueous droplets unacceptably compromised the heater lifetime. Because of this concern, the heater was prevented from contacting any liquids by positioning it outside the PCR mixture preparation stage of the fluidic device. At this location, a heater operating at the relevant current density has never been observed to fail during the course of a PCR run.
The on-chip temperature control system relied on the feedback from a temperature sensor (Omega CO1-Ta foil thermocouple) mounted on the backside of the chip. The correlation between the thermocouple reading and the temperature experienced by the PCR mixture droplet was established by placing another thermocouple on the front side of the chip sandwiched between the two plates of the EWD device and aligned to the backside thermoc ouple. Based on measurements, backside mounting necessitated a somewhat slower ramp-rate qPCR (overall ramp rate of 1.8oC/sec) so that the backside temperature measurement was consistent with the temperature in the droplet on the front side. This compares with 1.4oC/sec overall ramp rate for the benchtop ABI 7900HT.
Due to the complex requirements imposed by the fluidic and thermocycling aspects of the application, a custom - made fluorescent sensor was constructed. The sensor was mounted externally to the c hip and comprised the following components:
a. A 4.5 mW 532 nm collimated diode laser that was directly attached to the filter cube. Its power supply was controlled by a LabVIEW program.
b. A filter cube installed with three filters (532 ± 5 nm excitation filter, 544 nm dichroic mirror, and 555 nm long-pass emission filter) and two fiber couplers.
c. Two optical fibers: cube-to-device (0.22 NA, 550μm core) and cube-to-spectrometer (0.22 NA, 910μm core.) and
d. A CCD spectrometer.
Results
Effect of GuSCN and IPF Washes on PCR
One role of the IPF washes was to reduce the concentration of GuSCN in the PCR mixture. To evaluate the purification power achieved by on-chip washes, UV absorbance measurements were performed to determine the GuSCN concentration. In particular, rather than a traditional spectrometer, a Nano Drop spectrometer was employed. The key difference between the two is sample volume. For the former, it is generally required to fill a 1ml cuvette. In the latter case, a measurement is performed on a 1-2 μl droplet. With the NanoDrop spectrometer it was possible to generate an absorbance standard curve at 230nm peak absorption wavelength in the range 0.5 to 5mM GuSCN. As the GuSCN concentration increased, the peak absorption wavelength red shifted to about 260nm. Thus, a second standard curve was generated for GuSCN concentrations in the range 5-500mM. These standard curves are shown in Figure 5.

Figure 5: Absorbance-GuSCN standard curves for low and high concentration range (left and right respectively). Each concentration n=3. Both plots R2 >0.99.
Benchtop PCR experiments were performed to determine the GuSCN concentration range in the PCR mixture where qPCR starts to be inhibited. The amount of GuSCN added to the reaction mixture was gradually increased and then the resulting variation of the threshold cycle was observed. The GuSCN solutions were separately prepared by diluting Wash Buffer 1 in the miniMAG purification kit with nuclease -free water to the desired GuSCN concentration. The final GuSCN concentrations tested were 0.05, 5, 15.8, 25, 50, 158, 250, and 500mM. In addition, there were two controls: the positive control that did not contain GuSCN, and the No Template Control (NTC) that did not contain the IC DNA and GuSCN. Nuclease-free water was added to the controls to maintain the 25μl reaction volume. For each condition there were three replicates. The results showed that GuSCN started to impede PCR at 25-50mM concentrations in a reaction mix [24].
Next, in order to determine the number of sample washes required in the IPF purification steps, experiments were performed in which purification was performed on chip and PCR was performed on a benchtop instrument (ABI 7900HT). The following major steps were performed: First, the magnetic beads suspended in WB1 were actuated by an external magnet either directly to the EB (no wash) or to the WB3 (1 ~ 2 washes.) Secondly, the beads along with the GuSCN carryover were brought into contact with the EB. Third, with the magnet holding the beads in place, the eluent (i.e., EB contaminated with GuSCN) was aspirated from the device and subsequently analyzed with an absorbance measurement on a Nano Drop spectrophotometer.
Following the outlined procedure, absorbance measurements indicated that after 0, 1, 2 washes the average GuSCN concentrations in the eluent were 131.6, 28.4, 0.8mM, respectively. As plotted in Figure 6, the concentrations correspond to purification powers 38, 176, and 6124. In our experiments, we used a buffer in WB1 containing 5M GuSCN. Since GuSCN starts to impede PCR at 25 ~ 50mM in a reaction mix, then a purification power of at least 5M/25mM = 200 is needed.
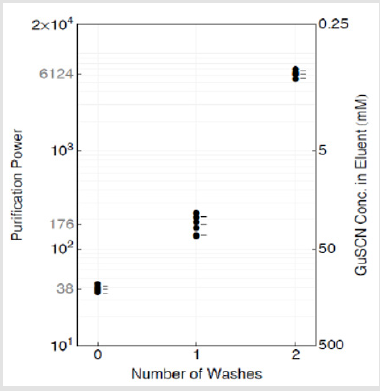
Figure 6: Purification power and GuSCN concentration in eluent vs. number of washes. Mean and 95% CI (t - distribution) are labelled as calculated from n=5, 6, 5 (0, 1, 2 washes respectively).
Results from On-Chip qPCR
Following the experimental purification method outlined above, on-chip qPCR was performed with the eluent resulting from 0, 1, and 2 IPF washes. Along with the NGC and NTC, each of the five types of tests was replicated three times. The obtained amplification plot is shown in Figure 7, and the threshold cycle data are tabulated in Table 1.
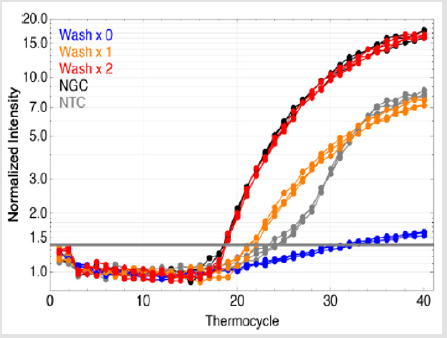
Figure 7: Threshold cycle vs. number of washes.
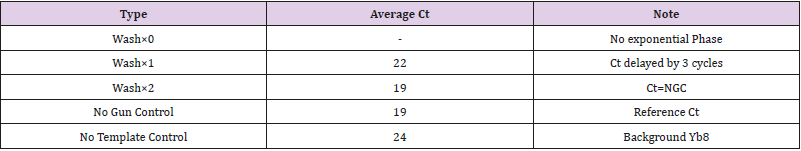
Table 1: Threshold cycle vs. number of IPF washes.
It can be seen in Figure 7 that the two-wash purification produced effectively the same amplification curve as the NGC. In other words, as estimated, a two-wash purification protocol is able to generate enough purification power to achieve uninhibited onchip qPCR. Single-wash purification, compared with NGC, resulted in a threshold cycle delayed by 3 cycles. Since each on-chip wash takes approximately twenty seconds, the extra wash does not significantly penalize the sample-to-answer time.
On-Chip Amplification Efficiency
The amplification efficiency, E, of on-chip qPCR as performed on this hybrid device was determined from the slope of the threshold cycle vs. initial copy number standard curve. On-chip qPCR was initiated after an Elution Buffer droplet was mixed with a master mix/assay/gDNA droplet. Three final DNA concentrations were tested: 0x, 1x, and 30x. For each concentration there were three replicates. Following the procedure described above, the obtained threshold cycle vs. initial copy number data are plotted in Figure 8.

Figure 8: On-chip qPCR amplification efficiency and Yb* background.
It can be seen that relative to a hypothetical case with 100% amplification efficiency (gray line), on-chip qPCR (black solid line) generates a lower threshold cycle when the starting copy number is high, and it generates a higher threshold cycle when the starting copy number is low. This behavior is symptomatic of low amplification efficiency. The actual efficiency value can be calculated from the slope of the solid line:
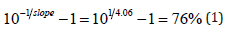
That is, on average 1.76 amplicons were generated per template per thermocycle, rather than the theoretical value of 2. Although the difference seems to be small, relative to a 100% efficient amplification, there will be 98% fewer amplicons after 30 thermocycles. Bench-top PCR amplification efficiency is typically 84%. The low efficiency suggests that on-chip qPCR requires further refinement.
The low amplification efficiency could be due to issues with the qPCR chemistry, the thermocycling profile, or a combination of both [24]. These effects can also explain the inability of one-wash purification to lead to uninhibited on-chip qPCR. By implication, one wash may become sufficient if the on-chip amplification efficiency can be improved to the bench-top level.
Nucleic Acid Retention
Nucleic acid retention was determined by performing all steps in the workflow, from the purification of gDNA from the PCR inhibitor solution by IPF washes to the detection of the Alu Yb8 target by qPCR. Good retention corresponds to a higher probability of having more copies of the targets in an eluent droplet, thereby improving the detection limit. Following off-chip binding of the gDNA to the magnetic silica beads, if 100% retention were achieved, the amount of the gDNA added to the Lysis/Binding Buffer would lead to 30x/8550 copies of Alu Yb8 in the final PCR mixture droplet.
After two IPF washes in WB3 were performed, the beads were transported to the EB reservoir. After a brief incubation, the gDNA was released from the beads. Subsequently, an eluent droplet and a master mix/qPCR assay droplet were dispensed from their respective reservoirs and then mixed together. Finally, the auxiliary system thermocycled the PCR mixture droplet and the resulting fluorescence was monitored. The present NA retention averaged 36%. As the experiment was structured, the obtained estimation of retention encompasses all of the losses that occurred throughout the workflow. Analysis has determined that the major contributor to poor NA retention was on-chip elution. This result is shown in Table 2 below, which indicates that that the loss due to elution was 3.4 times larger than the loss during IPF washing.

Table 2: Loss of nucleic acid retention during processing comparing On-Chip (“on”) and Off-Chip (“off”) Procedures.
Because elution is the step that bridges NA purification with EWD droplet-based PCR mixture preparation, the optimization of the elution protocol in the EB reservoir is challenging due to sometimes conflicting requirements. For example, the approach calls for eluting to a small volume of EB to improve the probability of having at least one copy of the target sequence in the eluent droplets. On the downside, the preference for NA to unbind from the silica beads during elution decreases with an increasing NA concentration in the eluent. Elution also is promoted by having an adequate degree of mixing between the beads and the EB. However, mixing is difficult in the current implementation because during elution the beads take up a substantial volume of the EB reservoir. Although the issue could be addressed by employing fewer beads, this solution limits the ability to use more beads to improve the probability of capturing the targets and to adjust the binding capacity.
Dynamic Range
The dynamic range is the range of initial template concentrations over which accurate Ct values are obtained. The floor of the dynamic range is determined by the threshold cycle of the No Template Control, i.e., the background human DNA contamination. The dynamic range determined from on-chip experiments was approximately 5x102, which ranges from 17 ~ 8.6 x 103 initial copies in the 92nl PCR mixture droplet [23]. From another point of view, after factoring in the 36% retention of DNA (from off-chip binding to on-chip purification and qPCR), prior to the binding step this range would correspond to 2.1 x 103 ~ 1.0 x 106 copies per ml of Lysis/Binding Buffer. In turn, 2.1 x 103 copies of Yb8 could be extracted from 0.6 somatic human cells, or in the case of a single copy per genome target amplifying at the same efficiency, 1.0 x 103 cells. In contrast, most qPCR reactions feature a dynamic range of 105~7, which ranges from 10 ~ 106~8 copies in a 10 ~ 25μl reaction volume. Hence, the dynamic range of on-chip qPCR at present lags by at least 200-fold. Fundamentally, this is a direct consequence of the low amplification efficiency.
Discussion
The PCR inhibiting concentration of GuSCN has been investigated by gradually increasing the amount of GuSCN added to the reaction mixture in benchtop PCR and then observing the resulting variation of the threshold cycle. It was found that GuSCN must be lowered from the initial 5M to less than 25 ~ 50mM. In other words, the purification power needed is at least 200.
The purification power gained by on-chip IPF washes was measured by correlating GuSCN concentration with the UV absorbance of the eluent generated by the purification stage of the fluidic device. After factoring in the purification power resulting from the preparation of the PCR mixture droplet, it was estimated that one- and two-wash purifications generated 352 and 12248 in purification power, respectively.
Although based on the above purification power estimations it was predicted that one wash would suffice, o n-chip qPCR reveals that in fact two IPF washes were needed. This may be due to the extra inhibitors introduced by the PCR stage taking up the inhibitor budget, and/or the overall inhibitor budget itself might become smaller because of a suboptimal thermocycling profile. Either of the two mechanisms will compromise the amplification efficiency.
The PCR amplification efficiency was found to be 76%. The value is indeed lower than the 84% obtained with the benchtop qPCR instrument, thereby supporting the aforementioned explanations. Additionally, the dynamic range of on-chip qPCR is shown to be approximately 17 ~ 8.6x103 copies of Yb8 in a 92nl PCR mixture droplet.
The average percentage of the analyte that is retained throughout the workflow is 36%. The experiment involved off-chip binding, onchip purification of the gDNA from the inhibitor solution, and the quantification of the retained Yb8 using on-chip qPCR. Although proof-of-principle demonstration was accomplished of a hybrid microfluidic device for sparse pathogen detection and analysis, further refinements will be required to improve NA retention and amplification efficiency.
Conclusion
The hybrid integration of fluidic Immiscible Phase Filtration (IPF) for purification and Digital Microfluidics (DM) for qPCR has been shown to be a potentially viable approach in performing timely molecular diagnostics for multiplex detection of sparse pathogens in large volume biofluids. Thus, as the first step towards such a sample-to-answer sparse pathogen detection device, the key result of this work is showing the feasibility of the IPF - DM integration.
The performance of a protype system has been characterized, including the purification power, amplification efficiency, NA retention, and the level of background contamination. Relative to a typical benchtop solid phase extraction procedure, a well-tuned IPF reagent system could attain the necessary degree of purification in appreciably fewer wash steps, thereby considerably reducing the purification time. Also, IPF is far more compatible with lowcost automation. In addition to purifying the NA, IPF also scales down the volume to what can be efficiently manipulated by DM. Thus, after elution DM could be used to automatically prepare the PCR reaction mixtures, which further reduces the hands-on time and the chance of contamination. Afterwards, either whole-chip thermocycling or temperature zone PCR can be employed. The latter in particular might substantially trim the PCR time. Notably, unlike the methods that rely on the magnetic beads to transport the droplets, DM can be scaled up to concurrently execute multiple multiplex qPCR reactions.
Acknowledgement
This work was supported in part by the National Science Foundation under grant NSF-CNS-11-35853.
References
- Sur K, McFall SM, Yeh ET, Jangam SR, Hayden MA, et al. (2010) Immiscible phase nucleic acid purification eliminates PCR inhibitors with a single pass of paramagnetic particles through a hydrophobic liquid. J Molecular Diagno 12: 620.
- Mercedes C Morales, Jeffrey D Zahn (2010) Droplet enhanced microfluidic-based DNA purification from bacterial lysates via phenol extraction. Microfluidics and Nanofluidics 9: 1041-1049.
- Lien KY, Liu CJ, Kuo PL, Lee GB (2009) Microfluidic system for detection of alpha-thalassemia-1 deletion using saliva samples. Anal Chem 81: 4502-4509.
- Sauer-Budge AF, Mirer P, Chatterjee A, Klapperich CM, Chargin D, et al. (2009) Low cost and manufacturable complete microTAS for detecting bacteria. Lab Chip 9: 2803-2810.
- Chen D, Mauk M, Qiu X, Liu C, Kim J, et al. (2010) An integrated, selfcontained microfluidic cassette for isolation, amplification, and detection of nucleic acids. Biomed Microdevices 12: 705-719.
- Easley CJ, Karlinsey JM, Bienvenue JM, Legendre LA, Roper MG, et al. (2006) A fully integrated microfluidic genetic analysis system with sample-in-answer-out capability. Proc Natl Acad Sci U. S. A 103: 19272- 19277.
- Thaitrong N, Toriello NM, Del Bueno N, Mathies RA (2009) Polymerase chain reaction-capillary electrophoresis genetic analysis microdevice with in-line affinity capture sample injection. Anal Chem 81: 1371-1377.
- Liu RH, Yang J, Lenigk R, Bonanno J, Grodzinski P (2004) Self-contained, fully integrated biochip for sample preparation, polymerase chain reaction amplification, and DNA microarray detection. Anal Chem 76: 1824-1831.
- Ramalingam N, Liu HB, Dai CC, Jiang Y, Wang H, et al. (2009) Real-time PCR array chip with capillary-driven sample loading and reactor sealing for point-of-care applications. Biomed Microdevices 11: 1007.
- Coelho B, Veigas B, Fortunato E, Martins R, Águas H, et al. (2017) Digital microfluidics for nucleic acid amplification. Sensors 17: 1495.
- Norian H, Field RM, Kymissis I, Shepard KL (2014) An integrated CMOS quantitative-polymerase-chain-reaction lab-on-chip for point-of-care diagnostics. Lab Chip 14: 4076-4084.
- Kim J, Gale BK (2008) Quantitative and qualitative analysis of a microfluidic DNA extraction system using a nanoporous AlOx membrane. Lab Chip 8: 1516.
- Oblath EA, Henley WH, Alarie JP, Ramsey M (2013) A microfluidic chip integrating DNA extraction and real-time PCR for the detection of bacteria in saliva. Lab Chip 13: 1325.
- Berry SM, Beebe DJ (2010) In 14th International conference on miniaturized systems for chemistry and life sciences. MicroTAS 1: 665.
- den Dulk RC, Schmidt KA, Gill R, Jongen JC, Prins MW (2010) In 14th International conference on miniaturized systems for chemistry and life sciences. Pp. 449.
- Lehmann U, Vandevyver C, Parashar V, Gijs M (2006) Droplet‐Based DNA Purification in a Magnetic Lab‐on‐a‐Chip. Angewandte Chemie- International Edition 45: 3062.
- Ohashi T, Kuyama H, Suzuki K, Nakamura S (2008) Control of aqueous droplets using magnetic and electrostatic forces. Analytica Chimica Acta 612 218-225.
- Pipper J, Zhang Y, Neuzil P, Hsieh TM (2008) Clockwork PCR including sample preparation. Angewandte Chemie-International Edition 47: 3900.
- Bienvenue JM, Legendre LA, Ferrance JP, Flanders JP (2010) Forensic Science International: Genetics 4: 178.
- Mahalanabis M, Do J, ALMuayad H, Zhang JY, Klapperich CM (2010) An integrated disposable device for DNA extraction and helicase dependent amplification. Biomed Microdevices 12: 353-359.
- Pollack MG, Eckhardt AE, Thwar P, Rouse J (2013) Method of ligating a nucleic acid.
- Hong Ren, Richard B Fair, Michael G Pollack, Edward J Shaughnessy (2002) Dynamics of electro-wetting droplet transport. Sensors and Actuators B: Chemical 87: 201.
- Hsu BN, Madison AC, Fair RB (2012) 16th International Conference on Miniaturized Systems for Chemistry and Life Sciences (MicroTAS 2012). Okinawa, Japan, pp. 830.
- Hsu BN (2014) Ph.D. Thesis. Duke University.
- Long Z, Shetty AM, Solomon MJ, Larson RG (2009) Fundamentals of magnet-actuated droplet manipulation on an open hydrophobic surface. Lab Chip 9: 1567-1575.
- Fair R (2007) Digital microfluidics: is a true lab-on-a-chip possible? Microfluidics and Nanofluidics 3: 245.
- Walker JA, Kilroy GE, Xing J, Shewale J, Sinha SK, et al. (2003) Human DNA quantitation using Alu element-based polymerase chain reaction. Analytical Biochemistry 315: 122-128.
- van der Horst EH, Leupold JH, Schubbert R, Ullrich A, Allgayer H (2004) TaqMan-based quantification of invasive cells in the chick embryo metastasis assay. Biotechniques 37: 940.
- Chern EC, Siefring S, Paar J, Doolittle M, Haugland RA (2011) Comparison of quantitative PCR assays for Escherichia coli targeting ribosomal RNA and single copy genes. Lett Appl Microbiol 52: 298-306.
- (2010) QuantiFast® Pathogen PCR +IC Handbook, Qiagen.
- (2011) TaqMan® Fast Advanced Master Mix Protocol. Applied Biosystems.
- https://www.biomerieux-usa.com/
- Lawyer FC, Stoel S, Saiki RK, Chang SY, Landre PA, et al. (1993) An efficient and economic enhancer mix for PCR. Genome Research 2: 275.
- McClernon DR, Ramsey E, StClair M (2007) Magnetic silica extraction for low-viremia human immunodeficiency virus Type 1 genotyping. J Clin Microbiol 45: 572-574.
