 Research Article
Research ArticleAbstract
Background: We have previously identified a naturally occurring alphavirus (M1) as an innovative oncolytic virus that targets tumors deficient in zinc-finger antiviral protein (ZAP). Although amount of studies reported that oncolytic viruses promote the anti-tumor activity of macrophages, the antiviral effect of macrophages and how it is affected by tumor is still largely unknown.
Methods: Using polycarbonate membrane inserts to establish co-culture model in which Murine macrophages Raw264.7 were co-cultured with itself or tumor cells (4T1 or B16F10). Flow cytometry, qRT-PCR, TCID50 assay, and Transcriptome sequencing were performed to determine the phenomenon and underlying mechanism of enhanced M1 virus replication in Tumor-conditioned macrophages.
Results: We first discovered that Oncolytic virus M1 can infect in and be eliminated by the macrophage. Furthermore, Tumor co-cultured treatment sensitized Raw264.7 macrophages to M1 replication and impeded M1 clearance from Raw 264.7. Finally, knock-down of HPGD in Raw264.7 showed similar M1-sensitive phenotype.
Conclusion: Tumor-derived factors inhibited antiviral immunity of Raw264.7 macrophages to M1 virus, which is associated with down-regulation of HPGD. This phenomenon may provide new insight into tumor selectivity of the M1 virus.
Keywords: Oncolytic Virus; tumor Associated Macrophage; Alphavirus M1; Breast Cancer; Melanoma
Abbreviations: ZAP: Zinc Finger Antiviral Protein; OVs: Oncolytic Viruses; FDA: Food and Drug Administration; ER: Endoplasmic Reticulum; T-Vec: Talimogene Laherparepvec; ER: Endoplasmic Reticulum
Introduction
Despite emerging advances in cancer treatment during the past decades, the increasing incidence of morbidity and mortality has made cancer the leading cause of death worldwide. Oncolytic viruses (OVs) have been used as an innovative biotherapeutic agent with the ability to specifically replicate in and kill cancer cells without harming normal cells [1]. In addition to direct cell killing by the virus, it has been demonstrated that a virus-induced immune response plays an important part in OV therapy [2,3]. Since OVs can destroy cancer cells via a mechanism distinct from conventional chemotherapy and radiotherapy, these viruses may be ideal for treating cancers that are nonresponsive to conventional treatment. To date, more than 10 species of OVs has been tested in pre-clinical and clinical trials and the milestone in this field is that the US Food and Drug Administration (FDA) approved talimogene laherparepvec (T-Vec), which is derived from HSV for treatment of melanoma patients, making it the first oncolytic virus approved for treating patients [4,5]. In spite of the evidence for oncolytic viruses killing cancer cells in vitro is unambiguous, the penetrance of oncolytic activity depends on various critical cellular barriers. For instance, the innate antiviral immunity in both normal tissue, especially the mononuclear phagocyte system, and tumor microenvironment.
As a key innate immunocyte, macrophage has a great threat in blocking the therapeutic dissemination of the oncolytic virus, which limits the efficacy of OVs as an anticancer agent. Some reports showed pharmaceutical inhibition or systemic depletion of macrophages could increase the administration efficacy and duration of OVs infection in tumor site [6, 7]. However, macrophages are a class of immune cells with extremely strong plasticity and macrophages of different phenotypes may have distinct effects on the virus. In some models of viral infection, M2-polarized macrophages promoted viral replication. For example, the massive replication of rhinovirus in chronic asthma is related to the inhibition of antiviral immunity by interleukin 10 (IL-10) secreted by M2 macrophages [8]. Linked to the similarity between Tumor-associated macrophages and M2 macrophages in the majority of tumor models [9,10], macrophages in the tumor microenvironment is not necessarily an obstacle to OVs. Our team has previously identified a strain of Getah-like alphavirus (M1), isolated from culicine mosquitoes in the Hainan province of China, as a naturally occurring oncolytic virus that targets zinc-finger antiviral protein (ZAP)-deficient cancer cells [11,12].
The genome of M1 is a positive single-strand RNA, which encodes several structural and nonstructural proteins. Importantly, we found that M1 causes prolonged and severe endoplasmic reticulum (ER) stress that in turn induced apoptosis in tumor cells and was nonpathogenic in nonhuman primates after multiple rounds of repeated i.v. injections [13]. After that, we have identified multiple signaling pathways associated with tumor cells killing effect of M1 and a series of related small molecule compounds as synergist [14-18]. However, a significant portion of cells in tumor mass is nontumorous [19], little is known about how these cells, especially the phagocytic macrophages usually occupying a large proportion of the tumor mass, affect antitumor effects of the M1 virus. Tumorassociated macrophage (TAM) was found to be a distinct phenotype compared with macrophages in normal tissue. Considering the tumor tropism of oncolytic virus M1, it is intriguing to examine if TAM has different responsiveness upon M1 infection. In this study, we investigated the infection and replication characteristics of M1 virus in macrophages and found that tumor-derived factors dampen the antiviral activity of Raw264.7 macrophages to M1 infection. These results imply that immuno-compromised of host innate immunity could be utilized by oncolytic M1 virus and also provided new insight into the tumor selectivity of M1 virus.
Materials and Methods
Cell Lines and Viruses
Cell lines were all purchased from American Type Culture Collection. Raw 264.7, 4T1, B16F10 were cultured at 37°C under 5% CO2 in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 100μg/ml streptomycin. THP-1 cells were cultured in RPMI 1640 supplemented with 10% FBS and stimulated with 50ng/ ml Phorbol-12-myristate-13-acetate (PMA, Sigma-Aldrich, USA) for 72h to differentiate as macrophages. All cell lines tested are negative for mycoplasma contamination. Production of Alphavirus M1 in this study was described previously [11]. M1-GFP is a recombinant derivative of M1 expressing jellyfish green fluorescent protein. Viruses were propagated in Vero cells (OPTISFM, Thermo Fisher, USA) and virus titers were determined by TCID50 in the BHK-21 cell line.
RNA Interference
Specific and nontargeting siRNAs were synthesized by Ribobio (Guangzhou, China). Cells were replaced with 10% fetal bovine serum DMEM (without penicillin/streptomycin). siRNAs were transfected using Lipofectamine RNAiMAX (Thermo Fisher) with OPTIMEM (Thermo Fisher).
Quantitative Reverse Transcription-Polymerase Chain Reaction
Total RNA was extracted using TRIzol (Life Technologies) reagent and reverse transcribed to cDNA with oligo (dT). Specific gene expression was quantified using SuperReal PreMix SYBR Green (FP204-02, TIANGEN, Beijing, China) on an Applied Biosystems 7500 Fast Real Time PCR system (Life Technologies). All genes were normalized to β-actin. Amplification primers (Thermo Fisher) are listed as follows:
Antibodies and Western Blot Analyses
Cells were lysed using M-PER Mammalian Protein Extraction Reagent (Thermo Scientific) and sodium dodecyl sulfate– polyacrylamide gel electrophoresis was performed. Antibodies used in this study are listed as follows: 15-PGDH (Novus, USA), phosphorylated STAT1(9167S, Cell Signaling Technology), STAT1 (9172S, Cell Signaling Technology), M1 virus E1 (produced by Beijing Protein Innovation, Beijing, China), β-actin (AP0063, Bioworld) , α-Tubulin (2125, Cell Signaling Technology).
Flow Cytometry
Cells were harvested and washed twice with cold PBS solution, immediately analyzed by flow cytometry (Becton–Dickinson, USA). M1-GFP infected cells were GFP+ and the infection rates were calculated relative to the total number of cells analyzed per sample (20,000 events) using CytExpert software.
Reagents and Recombinant Protein
Phorbol-12-myristate-13-acetate (PMA, Sigma-Aldrich, USA), Lipopolysaccharide (LPS, dissolved in ddH2O, Sigma-Aldrich, USA), SW033291 (Selleck, USA). Mouse IFN-γ, IL-4, IL-13 (Sino Biological, Beijing, China).
Statistical Analysis
Statistical analysis and graphs were made using GraphPad Prism (version 6.01, USA). Most of the data were analyzed by the Student t-test or one-way ANOVA with Dunnett’s tests for pairwise comparison. Bars show the mean ± SD of at least three independent repeat experiments.
Results
Interferon Pathway is Involved in the Clearance of Oncolytic Virus M1 by Macrophages
To explore whether M1 virus can infect and replicate in macrophage, mouse macrophage Raw264.7 and PMA-induced human macrophages THP-1 were infected with M1-GFP. The presence of green fluorescent protein (GFP) indicating that the M1 virus could enter macrophage and express the reporter gene inserted into the viral genome. As shown in Figure 1A, the expression of GFP protein in macrophages Raw.264.7 was observed at different time points after the oncolytic virus M1 infection, which suggested the replication and clearance of the virus in macrophages. In the two groups (10MOI, 1MOI) infected with high and medium titer M1 virus, the expression of GFP protein appeared earlier, but the clearance of GFP was also early, and the ratio of GFP positive cells after 48hpi (hours post-infection) significantly decreased, suggesting that the M1 virus in macrophages began to be cleared. In the experimental group infected with the low titer M1 virus (0.1 MOI), GFP expression was not observed until a relatively late time point (48 hpi). However, over time, there was a significant decrease in GFP expression at a later time point (72 hpi).
Similarly, we observed a similar phenomenon in PMAinduced human macrophage THP-1 (Figure 1B). To further study this phenomenon, we detected and calculated the fold change of intracellular virus genomic RNA expression by qRT-PCR (Figure 1C) and the supernatant or intracellular virus titers by TCID50 (Figure 1D). Consistent with the observation of Fluorescence microscope, the more M1 virus be used, the earlier it is cleared away from the culture system. These results indicate that macrophages are sensitive to M1 virus infection, and the replication of the virus at low titers is not significantly affected, and that macrophages can be activated to clear the virus after a certain degree of replication reaching a sufficiently high viral titer. To examine whether the interferon pathway is involved in this clearance process, we examined the expression of interferon in Raw264.7 cells before and after infection and found that infection and replication of M1 virus stimulated the expression of type I interferon (Figure 1E). After treatment with LPS and IFN-γ, it was detected that the M1 viral genomic RNA level and the supernatant virus titer in the macrophage Raw264.7 were decreasing (Figure 1F). These results have suggested that interferon is involved in the clearance of viruses by macrophages.
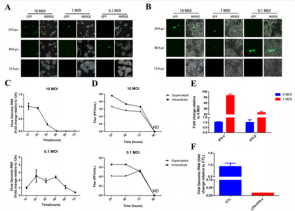
Figure 1: The containment and clearance of macrophage on the replication of oncolytic virus M1. A-B. Infection with oncolytic virus M1 expressing exogenous green fluorescent protein (GFP) at different multiplicity of infection (0.1, 1, 10)
a) Murine macrophage cell line Raw264.7 and
b) PMA induced Human macrophage cell line THP-1. At various time points after virus infection (24, 48, 72 hpi), cell morphology and GFP expression were observed using a fluorescence inverted microscope and photographed with a random field of view.
c) Semi-quantitative detection of viral genomic RNA in Raw264.7 cells using qRT-PCR at various time points post infection.
d) CCID50 method to detect the viral titer of the culture supernatant and intracellular (lysing Raw264.7 cells by liquid nitrogen freeze-thaw).
e) Medium titer (1 MOI) oncolytic virus M1 infected Raw264.7 cells, and the expression levels of interferon IFN-α, IFN-β and IFN-γ were detected using qRT-PCR.
f) IFN-γ (2.5 ng/ml) + LPS (200 ng/ml) treatment of Raw264.7 macrophages, compared with the untreated control, 24
h after mid-titer (1 MOI) oncolytic virus M1 infection. The amount of intracellular viral genomic RNA and viral titers in the supernatant. RNA Fold-expression was normalized to β-actin. ND: Non-Detectable.
Tumor Cells Can Attenuate the Clearance of Oncolytic Virus M1 By Macrophages in a Non-Contact Dependent Manner
We next sought to explore M1 replication in TAM-like macrophages. Raw264.7 cells pretreated with either self-conditioned medium or breast cancer cell 4T1 conditioned medium (CM) were infected with M1-GFP. To our surprise, Raw264.7 cells pretreated with 4T1CM were seemingly more sensitive to M1 virus infection (Figure 2A). This suggested that substances secreted by tumor cells may impaire the antiviral activity of macrophages against oncolytic virus M1. To further mimic tumor microenvironment and exclusively observe M1 replication in macrophage other than in tumor cells, we cultured RAW264.7 together with 4T1 cells (4T1CC), mouse melanoma cells B16F10 (B16F10CC) or Raw264.7 itself (control cells, RAWCC) in upper well, using polycarbonate membrane inserts to avoid direct contact between the two cell types. Similarly, the cell number of GFP+were significantly higher in Raw264.7 cells co-cultivation pretreated with 4T1 or B16F10 than in Raw264.7 cells co-cultivation pretreated with themselves (Raw264.7) (Figure 2B & 2C), suggested that the non-contact effect of tumor cells increases the containment of macrophage to oncolytic virus M1. To analyze the replication of the virus, qRT-PCR was used to detect the expression level of intracellular viral genomic RNA, and the TCID50 method was used to detect the amount of virus in the supernatant. The experimental results showed that after viral infection (24hpi and 48hpi), both viral genomic RNA (Figure 2D) in tumor cell-cultured macrophage Raw264.7 cells and viral titer in the extracellular supernatant (Figure 2E & 2F) were significantly increased compared with the self-cultivation control group. In addition, it can be seen from the results that after 48 hours of infection, both the amount of viral genomic RNA expression (Figure 2D) and the viral titer in the extracellular supernatant (Figure 2F) were significantly reduced in the control group, which did not occur in the test group.
Although the expression of viral genomic RNA (Figure 2D) in the co-cultivation pretreated macrophage Raw264.7 cells also decreased at 48hpi comparing with 24hpi, the trend was weakened comparing with the control group. The virus titer in the supernatant was not found a significant reduction at 48hpi comparing with 24hpi, in fact, there was even a certain increase (Figure 2E & 2F). In view of our previous finding that more virus amount can lead to earlier onset of clearance, the postponed clearance in the test group (Figures 2D &2F) suggested that tumor cells may impair macrophage clearance of oncolytic virus M1, even though cannot rule out the possibility of promotion of the viral replication in macrophages by cancer cell. By comparing the occurrence order between the intracellular viral genomic RNA decease and extracellular virus titer reduction, it was revealed that intracellular clearance may precede extracellular clearance in our macrophagecancer cell interaction system.
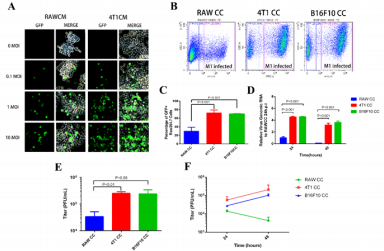
Figure 2: Co-culture pretreatment by cancer cells impairs the virus clearance of oncolytic virus M1 by macrophages Raw264.7.
a) Pretreatment of Raw264.7 cells with self-conditioned medium (RAWCM) and 4T1 conditioned medium (4T1CM) for 48h, infected with different titer oncolytic virus M1, and observed GFP+ cells at 24hpi using fluorescence phase contrast microscope to evaluate the replication of tumor virus M1.
b) Co-culture with different cells (RAWCC RAW264.7; test group 4T1CC using 4T1; test group B16F10CC using B16F10) in upper chamber and with RAW264.7 in lower chamber of the transwell system, co-cultured for 72h and then infected Raw264.7 with 10MOI oncolytic virus M1, Raw264.7 cells were harvested at 24 hpi, and the ratio of GFP-positive cells was measured by flow cytometry.
c) RAW264.7 cells in the lower chamber as described above were infected with 1 MOI oncolytic virus M1. 24 h and 48 h after infection, the cells were harvested for qRT-PCR to quantify the amount of viral genomic RNA in the cells.
d) Detection of virus titer in Raw264.7 cell supernatant using TCID50 method at different time points (24 hpi and 48 hpi) after infection with oncolytic virus M1 at different titers (10 MOI, panel E; or 1 MOI, panel
Tumor Cells Co-Culture Pretreatment Did Not Impair Interferon Pathway Activation After Oncolytic Virus M1 Infection
As previously reported [20], tumor-derived exosomes inhibit the production of IFN-β in influenza-infected macrophages, which impaired the clearance activity of macrophage. In order to illustrate the mechanism of impairment, we first examine the type1 IFN (IFN-α, IFN-β) and type-2 IFN (IFN-γ) mRNA expression before and after M1 infection in Raw264.7 cells under different co-culture pretreatment conditions (Figure 3A). It was found that compared with the self-cultivation control group, the tumor cell co-culture pretreatment promoted the expression of various types of interferon infection. Among them, the increase of type I interferon is more significant, especially the raw264.7 under 4T1 co-culture pretreatment (Figure 3A). Subsequently, by detecting the phosphorylation activation of STAT1 and the expression of interferon-stimulated genes (ISGs) CXCL10 and ISG15 downstream of the type I IFN pathway. It was consistently found that the M1 virus caused the activation of the type I interferon pathway in Raw264.7 cells. The co-culture pretreatment of tumor cells does not only inhibit the interferon cell pathway but activate it more effectively (Figure 3B & 3C). These results suggest that cancer cells such as 4T1 and B16F10 are likely to reduce the clearance of M1 by macrophages by affecting other changes than the interferon pathway.
Figure 3: Cancer cell co-culture pretreatment does not impair the activation of interferon-related pathways in Raw264.7 cells by oncolytic virus M1.
a) Detection of type I and type II interferon IFN mRNA expression using quantitative RT-PCR after infection with 1MOI oncolytic virus M1 for 24 hours in different co-culture pretreated Raw264.7.
b) Western blot was used to detect protein expression and phosphorylation activation of STAT1 after 24 hours of MOI oncolytic virus M1 infection in Raw264.7 cells pretreated with different co-cultures. qRT-PCR was used to detect mRNA of expression of CXCL10 and ISG15, two genes of interferon-stimulated genes downstream of the type I interferon pathway (ISG), after 24 hours of MOI oncolytic virus M1 infection in Raw264.7 cells pretreated with different co-cultures.
Down-regulation of HPGD in Tumor Co-cultured Raw264.7 is associated with Enhanced M1 Sensitivity
To determine the underlying mechanism of impaired M1 clearance by macrophages, we used Raw264.7 cells co-cultivation pre-treated with themselves as a control group (RAWCC), and co-cultivation pre-treated Raw264.7 cells with cancer cells 4T1 and B16F10 as two different test groups (4T1CC and B16F10CC). All three groups were subjected to transcriptome sequencing by BGISEQ-500 platform. In order to find out the more common macrophage gene expression patterns caused by different tumor cells (4T1CC and B16F10CC), we first analyzed the genes with the same trend expression changes in macrophage RAW264.7 in different experimental groups. The Log2FoldChange (LogFC) values obtained by comparing the FPKM values of the genes in each group were sorted and found that HPGD gene was greatly down-regulated (LogFC < -5) in both 4T1CC and B16F10CC compare to the RAWCC (Green dot in the lower left box in Figure 4A. Relatively, up to 15 genes were significantly up-regulated (LogFC >5) in 4T1CC and B16F10CC (Red dots in the upper right box in Figure 4A. Therefore, we first concentrated on the down-regulated HPGD gene. Further, we confirmed the results of transcriptome sequencing by qRT-PCR.
The HPGD gene expression in Raw264.7 cells were significantly down-regulated after co-culture with 4T1 and B16F10 tumor cells (Figure 4B). To investigate whether down-regulation of HPGD gene expression is associated with increased replication of oncolytic virus M1 in Raw264.7 cells, we used RNA interference to reduce the expression of HPGD in Raw264.7 cells and observe M1 expression. After successful knockdown of HPGD at mRNA (Figure 4C) and protein (Figure 4D) levels, the replication of oncolytic virus M1 was significantly increased (Figure 4E & 4F). Pharmaceutically inhibition of 15-PGDH (15-hydroxyprostaglandin dehydrogenase), which is encoded by HPGD also showed a similar phenomenon (Figure 4G & 4H). These results suggest that cancer cells such as 4T1 or B16F10 might dampen the antiviral clearance activity of Raw264.7 macrophages via down-regulation of HPGD.
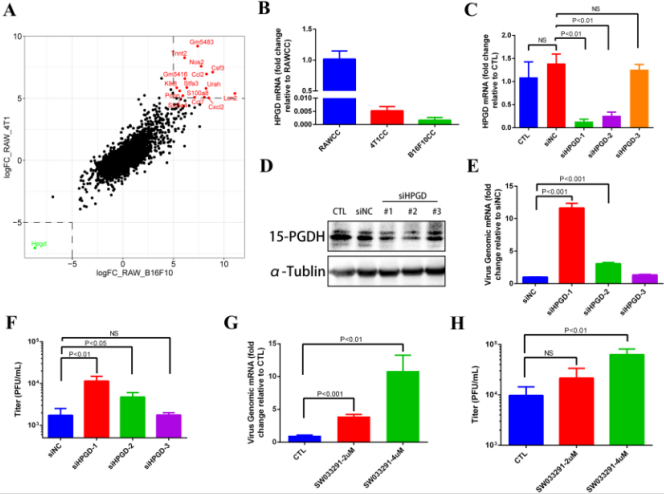
Figure 4: Down-regulated expression of HPGD gene in tumor co-culture pretreatment Raw264.7 cells and its effect on macrophage virus clearance.
a) Genes Log Fold Change of RPKM values in 4T1CC (Y-axis) or B16F10CC (X-axis) relative to the Control RAWCC.
b) HPGD mRNA expression fold change in 4T1CC or B16F10CC relative to RAWCC.
c) RNAi efficiency of three siRNA fragments examined by qRT-PCR and Western Blot.
d) M1 virus replication in Raw264.7 knock-down of HPGD (1 MOI, 24 hpi) detected by qRT-PCR and TCID50.
e) M1 virus replication in Raw264.7 (1 MOI, 24 hpi) pretreated with 15-PGDH inhibitor SW033291 for 24h detected by qRTPCR and TCID50.
Discussion
Our results reveal the in vitro dynamic characteristic of oncolytic virus M1 replication and clearance in macrophages. Moreover, mouse breast cancers cell 4T1 and melanoma cells B16F10 restrained the antiviral activity of Raw264.7 mouse macrophage to M1 infection and the underlying mechanism is associated with the significant down-regulation of HPGD in Tumor co-cultured Raw264.7. As previously reported, the tumor selectivity of the oncolytic virus is principally based on the genetic abnormalities of malignant cells, including aberrant oncogenic signaling, innate immune defects, and tumor-specific receptors [21- 23]. Since the tumor microenvironment (TME) consists of various cell types (endothelial cells, fibroblasts, immune cells, etc.) that are nontumorous and affected by tumor cells [19], it has inevitable effects on therapeutic efficacy, for example the environment mediated drug resistance protects tumor cells from apoptosis induced by either chemotherapy, radiotherapy or receptormediated cell death [24]. For oncolytic virotherapy, the situation might be different because the TME, which commonly considered as “immunosuppressive” influenced by tumor cells, is possibly a suitable habitat for OVs relative to normal tissue. To verify this possibility, we used in vitro non-contact cell co-cultured model to partially simulate the stimulus received by macrophage in TME and identified a phenotypic transition occur in tumor co-cultured Raw264.7 macrophages which are more sensitive to oncolytic virus M1 infection and replication. These results help us to uncover the mechanism of tumor selectivity of the M1 virus from macrophages perspective other than tumor cells and finally increase the response rate of M1 in future clinical trials. 15-hydroxyprostaglandin dehydrogenase (15-PGDH), encoded by HPGD, is a key enzyme responsible for inactivation of PGE2 in tissue and has a tumor suppressor function in multiple cancer types including colon, lung, and breast cancers [25-27].
Therefore it is frequently low expression in tumor cells, and together with the overexpression of cyclooxygenase-2 (COX-2), a higher amount of PGE2 are secreted and persevered in TME, which is often associated with tumor-induced immune suppression [28,29].Through high-throughput transcriptome sequencing, we notice that HPGD was most significantly down-regulated in both tumor co-cultured groups (4T1CC, B16F10CC). Knockdown of HPGD in Raw264.7 significantly increased M1 virus replication, indicating that 15-PGDH may function as an antiviral factor in Raw264.7. According to previous reports and our study on M1 infection in M2-polarized macrophages (data not shown), the exact mechanism that down-regulation of HPGD hinder Raw264.7 macrophages to eliminate M1 virus might be correlated with M2 macrophage polarization mediated by 15-PGDH deficiency and PGE2 accumulation [30,31].
References
- Russell SJ, KW Peng, JC Bell (2012) Oncolytic virotherapy. Nat Biotechnol 30(7): 658-670.
- Oh E, Choi IK, Hong J, Yun CO (2017) Oncolytic adenovirus coexpressing interleukin-12 and decorin overcomes Treg-mediated immunosuppression inducing potent antitumor effects in a weakly immunogenic tumor model. Oncotarget 8(3): 4730-4746.
- Kim JH, Oh JY, Park BH, Lee DE, Kim JS, et al. (2006) Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther 14(3): 361-370.
- Johnson DB, I Puzanov, MC Kelley (2015) Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy 7(6): 611-619.
- Berkey SE, SH Thorne, DL Bartlett (2017) Oncolytic Virotherapy and the Tumor Microenvironment. Adv Exp Med Biol 1036: 157-172.
- Fulci G, Breymann L, Gianni D, Kurozomi K, Rhee SS, et al. (2006) Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci USA 103(34): 12873-12878.
- Shashkova EV, Doronin K, Senac JS, Barry MA (2008) Macrophage depletion combined with anticoagulant therapy increases therapeutic window of systemic treatment with oncolytic adenovirus. Cancer Res 68(14): 5896-904.
- Zdrenghea MT, Makrinioti H, Muresan A, Johnston SL, Stanciu LA (2015) The role of macrophage IL-10/innate IFN interplay during virus-induced asthma 25(1): 33-49.
- Chanmee T, Chanmee T, Ontong P, Konno K, Itano N (2014) Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 6(3): 1670-1690.
- Goswami KK, Ghosh T, Ghosh S, Sarkar M1, Bose A, et al. (2017) Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol 316: 1-10.
- Hu J, XF Cai, G Yan (2009) Alphavirus M1 induces apoptosis of malignant glioma cells via downregulation and nucleolar translocation of p21WAF1/CIP1 protein. Cell Cycle 8(20): 3328-3339.
- Lin Y, Haipeng Zhang, Jiankai Liang, Kai Li, Wenbo Zhu, et al. (2014) Identification and characterization of alphavirus M1 as a selective oncolytic virus targeting ZAP-defective human cancers. Proc Natl Acad Sci USA 111(42): 4504-4512.
- Zhang H, Yuan Lin, Kai Li, Jiankai Liang, Xiao Xiao, et al. (2016) Naturally Existing Oncolytic Virus M1 Is Nonpathogenic for the Nonhuman Primates After Multiple Rounds of Repeated Intravenous Injections. Hum Gene Ther 27(9): 700-711.
- Li K, Zhang H, Qiu J, Lin Y, Liang J, et al. (2016) Activation of Cyclic Adenosine Monophosphate Pathway Increases the Sensitivity of Cancer Cells to the Oncolytic Virus M1. Mol Ther 24(1): 156-165.
- Cai J, Lin Y, Zhang H, Liang J, Tan Y, et al. (2017) Selective replication of oncolytic virus M1 results in a bystander killing effect that is potentiated by Smac mimetics. Proc Natl Acad Sci USA 114(26): 6812-6817.
- Liang J, Li Guo, Kai Li, Xiao Xiao, Wenbo Zhu, et al. (2018) Inhibition of the mevalonate pathway enhances cancer cell oncolysis mediated by M1 virus. Nat Commun. 9(1): 1524.
- Li K, Hu C, Xing F, Gao M, Liang J, et al. (2018) Deficiency of the IRE1alphaAutophagy Axis Enhances the Antitumor Effects of the Oncolytic Virus M1. J Virol 92(6).
- Zhang H, Li K, Lin Y, Xing F, Xiao X, et al. (2017) Targeting VCP enhances anticancer activity of oncolytic virus M1 in hepatocellular carcinoma. Sci Transl Med 9: (404).
- Wu T, Y Dai (2017) Tsumor microenvironment and therapeutic response. Cancer Letters. 387: 61-68.
- Gao L, Lin Wang, Tong Dai, Ke Jin, Zhengkui Zhang, et al. (2018) Tumorderived exosomes antagonize innate antiviral immunity. Nat Immunol 19(3): 233-245.
- Angelova AL, Geletneky K, Nüesch JP, Rommelaere J, et al. (2015) Tumor Selectivity of Oncolytic Parvoviruses: From in vitro and Animal Models to Cancer Patients. Front Bioeng Biotechnol 3: 55.
- Komatsu Y, Christian SL, Ho N, Pongnopparat T, Licursi M, et al. (2015) Oncogenic Ras inhibits IRF1 to promote viral oncolysis. Oncogene 34(30): 3985-3993.
- Ahtiainen L, Cristina Mirantes, Tiina Jahkola, Sophie Escutenaire, Iulia Diaconu, et al. (2010) Defects in innate immunity render breast cancer initiating cells permissive to oncolytic adenovirus. Plos One 5(11): 13859.
- Meads MB, RA Gatenby, WS Dalton (2009) Environment-mediated drug resistance: a major contributor to minimal residual disease. Nature Reviews Cancer 9: 665.
- Tai HH, M Tong, Y Ding (2007) 15-hydroxyprostaglandin dehydrogenase (15-PGDH) and lung cancer. Prostaglandins Other Lipid Mediat 83(3): 203-208.
- Wolf I, O Kelly J, Rubinek T, Tong M, Nguyen A, et al. (2006) 15-hydroxyprostaglandin dehydrogenase is a tumor suppressor of human breast cancer. Cancer Res 66(15): 7818-7823.
- Dumont P, Alix Berton, Nathalie Nagy, Flavienne Sandras, Sandrine Tinton, et al. (2008) Expression of galectin-3 in the tumor immune response in colon cancer Lab Invest 88(8): 896-906.
- Kim R, M Emi, K Tanabe (2006) Cancer immunosuppression and autoimmune disease: beyond immunosuppressive networks for tumour immunity. Immunology 119(2): 254-564.
- Sinha P, Clements VK, Fulton AM, Ostrand Rosenberg S (2007) Prostaglandin E2 promotes tumor progression by inducing myeloidderived suppressor cells. Cancer Res 67(9): 4507-4513.
- Yamamichi K, Fukuda T, Sanui T, Toyoda K, Tanaka U, et al. (2017) Amelogenin induces M2 macrophage polarisation via PGE2/cAMP signalling pathway. Arch Oral Biol 83: 241-251.
