 Review Article
Review ArticleAbstract
Organophosphorus nerve agents, such as soman, induce a cholinergic crisis by inhibiting the enzyme acetylcholinesterase throughout the nervous system. While current medical countermeasures effectively mitigate peripheral effects, the brain is vulnerable to severe damage as sustained seizure activity is refractory to treatment. Because adenosine (ADO) has profound inhibitory effects in the brain, stimulation of A1 adenosine receptors has been hypothesized to be an effective therapeutic strategy against nerve agents. The Netherlands Organization for Applied Scientific Research (TNO) was the first to test that hypothesis in 1998 and demonstrated some success. However, TNO discontinued adenosine research in the early 2000s because of adenosine’s cardiovascular side effects. We rekindled adenosine-nerve agent research in 2012 and tested novel treatment strategies using the A1 adenosine receptor agonist N6-cyclopentyladenosine (CPA). We demonstrated that CPA injected into the brain or periphery at high doses was highly neuroprotective against soman. Results strongly indicated that CPA prevented/terminated seizure via pre- and post-synaptic neuronal mechanisms. Data also suggested that immediate CPA treatment may have protected acetylcholinesterase from soman inhibition. Additionally, the hypothermia that develops after ADO treatment may also enhance neuroprotection and survivability after exposure to nerve agents. Further research is needed to better understand adenosine’s promising neuroprotective mechanisms and therapeutic potential as novel medical countermeasure for nerve agent intoxication. Here is a review of current research in progress.
Keywords: Adenosine Agonist; Nerve Agent; Neuroprotection; Anti-Seizure; Soman
Abbreviations: ADO: adenosine; OP: Organophosphorus; CWNAs: Chemical Warfare Nerve Agents; AChE: Acetylcholinesterase; NT: Neurotransmitter; CNS: Central Nervous System; NMDA: N-Methyl-D-Aspartic Acid; BBB: Blood Brain Barrier; ARs: Adenosine Receptors; BF: Basal Forebrain; IM: Intramuscularly; NECA: 5’-N Ethylcarboxamidoadenosine; ICV: Intracerebroventricularly; 8-SPT: 8-(p-Sulfophenyl)Theophylline; CPA: N6-cyclopentyladenosine
Introduction
Organophosphorus (OP) chemical warfare nerve agents (CWNAs), such as soman, sarin, tabun, and VX, irreversibly inhibit acetylcholinesterase (AChE), the enzyme responsible for hydrolyzing the neurotransmitter (NT) acetylcholine (ACh) in cholinergic synapses and neuromuscular junctions [1]. Following exposure to CWNAs, the earliest reported neurochemical change in the central nervous system (CNS) is an immediate inhibition of AChE activities and subsequent elevation of brain ACh levels. Elevation in central extracellular ACh leads to widespread neural activation and seizure generation [2]. Following sustained seizure activity, the cholinergic crisis initiates changes in the levels of excitatory (glutamate) and inhibitory (γ-Aminobutyric acid, GABA) amino acid transmitters [2-9]. Numerous inhibitory compounds with different pharmacologic mechanisms have been investigated to combat this toxic sequelae. Drugs that block ACh receptors (e.g., atropine and scopolamine), enhance the effect of γ-amino-butyric acid (GABAA) receptors (diazepam and midazolam) or antagonize N-methyl-D-aspartic acid (NMDA) glutamatergic receptors (MK801) are efficacious when administered soon after CWNA exposure [10,11]. However, after a period of sustained seizure activity, the CNS often becomes refractory to these treatment strategies, and seizures cannot be terminated [10,12].
In addition to inhibitory compounds, current treatment strategies also include oximes such as pyridine-2-aldoximemethylchloride (2-PAM) or obidoxime to reactivate the AChE activities and suppress any elevation in ACh levels. While effective at reactivating AChE in the periphery, currently available oximes do not cross the blood brain barrier (BBB) because of their quaternary structure and, therefore, leave the CNS unprotected from seizure [13]. Moreover, some CWNAs such as soman and its bound AChE will rapidly undergo a process known as ageing where it becomes resistant to oxime reactivation and seizure will continue [14]. Because current inhibitory and oxime-based strategies are unable to control seizure activity when treatment is delayed, it is essential that patients are treated as quickly as possible with these types of drugs so that seizures are suppressed. Otherwise, unmitigated hyperactive neuronal activity will cause significant neuropathology and possibly death [15]. Since a CWNA attack may overwhelm the first responders’ capacity to quickly treat all victims, new medical countermeasures capable of terminating sustained seizure activity need to be developed. Towards that objective, recent studies in our laboratory have investigated adenosine (ADO) receptors (ARs) and their agonists in small animal models for their anti-seizure and neuroprotective properties. This paper will review the promising findings from these studies and discuss future directions to research and develop this class of compounds for possible clinical applications.
Endogenous Adenosine and its Physiologic Effects
ADO is a purine nucleoside that participates in multiple peripheral and central physiologic functions. It is released during normal metabolic activity into the extracellular space where it acts on ARs [16]. ADO in the CNS also plays a role in modulating inflammatory processes. While the specific mechanisms are not fully understood, studies have shown that ADO can protect against neuronal damage in cases of ischemia, epileptic seizures, and CWNA exposure [17-20]. It is hypothesized that ADO’s neuroprotective benefits are attributable to its primary role in the CNS as an inhibitory neuromodulator. ADO modulates cellular activity by stimulating specific AR subtypes which are classified according to their effect on adenylyl cyclase; A1 inhibits activity via Gαi proteins, and A2A enhances activity via Gαs proteins [21]. ADO’s most notable physiologic effect occurs with the stimulation of A1 ARs in the brain. Activation of central A1 ARs elicits a profound inhibitory effect on neuronal excitability. Specifically, ADO suppresses neuronal activity by presynaptic inhibition of neurotransmitter release and post-synaptic hyperpolarization [16-22].
A1 ARs are distributed throughout numerous brain structures including the cortex, thalamus, and critical cholinergic centers: the basal forebrain (BF), hippocampus, and striatum [19-23]. In addition to the brain, A1 ARs are also widely distributed throughout the periphery. A1 ARs have been detected in the heart, aorta, liver, kidney, eye, and bladder [24]. In the periphery, A1 ARs primarily decrease heart rate and blood pressure [25]. A2A ARs are also located in both the periphery and brain, but are expressed to a much lesser extent. In the CNS, A2A ARs are primarily expressed in the basal ganglia, and less so in hippocampus, cerebral cortex, nucleus tractus solitarius, and motor nerve terminals [26,27]. Activation of A2A ARs in the brain enhances the release of neurotransmitters and may promote GABAergic signaling [28]. Peripheral A2A ARs are located primarily in the vasculature and decrease blood pressure by mediating vasodilation [25,29].
Neuroprotective Efficacy of Systemically Administered Adenosine
Research has shown that exogenously administered ADO provides neuroprotection from various traumas, including hypoxia and ischemia [30-32]. Data suggest that ADO’s protective mechanism involves the partial neutralization of neuronal Ca++ overload that leads to cell death [31]. ADO’s inhibitory effect on neuronal excitability has also been exploited for the treatment of drug-resistant epilepsy [33-36]. The A1 AR’s distribution throughout the CNS, including CWNA-affected cholinergic centers, makes it an attractive therapeutic target for new medical countermeasures to OP nerve agents [19]. van Helden and his associates from The Netherlands Organization for Applied Scientific Research (TNO, Netherlands) pioneered the use of an ADO agonist (N6-cyclopentyladenosine, CPA) as an alternative treatment of nerve agent poisoning [20]. In one initial experiment, Van Helden et al. [20] intramuscularly (IM) administered the ADO agonist 5’-N-Ethylcarboxamidoadenosine (NECA) or CPA into rats following exposure to soman. The ADO agonists prevented or delayed the cholinergic crisis and decreased soman’s lethality. Van Helden et al. [20] and his colleagues followed up that promising study with experiments that further investigated ADO’s efficacy against CWNAs. In one study, systemic CPA at 1-2 mg/kg was shown to be efficacious in rats exposed to tabun or sarin, but not to VX [37]. The results from a subsequent TNO study suggested that CPA protected central AChE by limiting the nerve agent’s distribution throughout the body [38]. To further investigate the link between CPA’s cardiovascular side effects and its neuroprotective mechanism, Joosen et al. [38,39] correlated sarin’s cholinergic symptoms with CPA-induced bradycardia and hypotension [39]. Because a peripherally acting A1 AR antagonist counteracted CPA’s cardiovascular effect and its neuroprotective efficacy, the authors concluded that cardiovascular depression was the source of therapeutic efficacy.
TNO subsequently discontinued their ADO research, theorizing that systemically administered ADO analogs could not reach the brain at sufficient concentrations to combat central ACh accumulation, and that CPA’s neuroprotection was a result of less nerve agent circulating to the brain because of ADO’s depression of cardiac output [38,39]. In 2012, we rekindled the idea of utilizing ADO agonists for CWNA neuroprotection. Although peripheral AR stimulation does have cardiovascular side effects, we believed that ADO’s profound central pre- and post-synaptic inhibitory effects warranted further investigation considering the limitations of currently available CWNA neuroprotectants. It was theorized that if ADO proved to be highly effective, either novel agonists or delivery methods could be developed to exploit ADO’s central benefits while minimizing peripheral side effects. Research began with direct brain injections of CPA to study the true neuroprotective efficacy of central AR stimulation. The results from that study demonstrated that CPA does have a direct effect on central neuronal activity that provides neuroprotection against CWNAs [40-42]. Subsequent studies transitioned to systemic CPA injections at significantly greater doses than what was tested at TNO. The results from these studies are described below.
Neuroprotective Efficacy of CPA Delivered Directly to the Brain
To isolate the neuroprotective efficacy of central A1 AR stimulation after exposure to CWNA, we conducted a study that administered CPA at 700µg intracerebroventricularly (ICV) or injected CPA at 350 µg directly into the cholinergic BF area 1 minute after a seizure-inducing dose of soman (180µg/kg, SC) in a rat model [40]. The results demonstrated that both widespread A1 AR stimulation via ICV CPA and focal BF A1 AR stimulation prevented the onset of soman-induced seizure and protected the brain from the development of pathology. After administration of CPA in both ICV and BF, neuronal activity and EEG power were immediately suppressed. Because the development of neuropathology closely correlates with sustained hyperactive brain activity, neuroprotection can be presumed to be provided by CPA’s direct effect on neuronal activity, and therefore, CPA’s anti-seizure effect was centrally mediated (vs. peripheral/cardiovascular). To further investigate the neuroprotective capacity of central AR stimulation, we assessed CPA’s ability to terminate soman-induced seizure well after its onset [42]. Twenty minutes after seizure induced by soman was detected via EEG, CPA was delivered ICV at 700µg. The results showed that CPA could terminate sustained seizure activity for 67% of the animals. Within approximately 3.5 hours of treatment, the hyperactive EEG activity was suppressed to low amplitude and frequency. Even though CPA via ICV did not demonstrate 100% efficacy, it should be noted that half of the rats that were unresponsive to treatment (2 of 4) died within 10 minutes of treatment, a period of time that is too short for CPA to act on the A1 ARs and to terminate seizure. Five of the 8 rats that were responsive and experienced seizure termination survived until the study’s endpoint of 48 hours. CPA’s efficacy in terminating sustained seizure activity suggests that treatment inhibited neuronal excitotoxic activity via post-synaptic effects. Better efficacy can be expected with greater centrally administered CPA doses. These promising results support the idea that CPA or other ADO agonists could greatly benefit the victims of a CWNA attack.
Development of Clinically Relevant Adenosine Treatment Regimens
Although ICV delivery of CPA mitigated much of the cardiovascular side effects in our studies, direct brain injections are not practical in the field or clinic settings. Systemic administration strategies are needed, however, peripheral injection of an ADO agonist may exacerbate the CWNA-induced peripheral cholinergic crisis and negatively affect survival. CPA’s neuroprotective benefits might possibly be harnessed without negative peripheral side effects by co-administering a BBB impermeable AR antagonist. Peripheral AR antagonism would minimize CPA’s cardiac side effects while maintaining its positive central effects. One such AR antagonist is 8-(p-sulfophenyl) theophylline (8-SPT), which has been shown to counteract AR-induced cardiovascular effects at 50mg/kg in a rat model [43]. Joosen et al. [39] used a similar approach to investigate the correlation between CPA’s cardiovascular effect and neuroprotection. In that study, a low dose of CPA (0.05mg/kg, IM) was co-administered with 8-SPT at 20mg/kg after sarin exposure. That treatment regimen displayed a marked lack of neuroprotective efficacy. We theorized that the dose of CPA was too low to reach and agonize central A1 AR receptors. Therefore, CPA (50-55mg/kg, IP) and 8-SPT (50mg/kg, IP) were given in combination 1 minute after a seizure-inducing dose of soman in our study. The results showed that systemically delivered CPA with 8-SPT effectively prevented seizure after soman exposure in all rats. Moreover, systemically administered treatment did not exacerbate soman’s peripheral effects or increase lethality. Survival rates between treatment and control groups were not statistically significant. No quantitative cardiovascular measures were obtained because of complications with instrumentation. However, qualitative assessment of mucus membranes and tissue coloring suggested that blood circulation was adequately maintained. The demonstration that seizures were consistently prevented by high doses of systemically administered CPA was an exciting finding and suggested that peripheral side effects may not be a definitive obstacle for ADO’s clinical application.
Adenosine’s Effect on Extracellular Neurotransmitter Levels
Since CWNAs initiate and maintain their central toxicity by elevating extracellular excitatory neurotransmitter levels, intense pre-synaptic inhibition could be an effective strategy to suppress seizure activity and provide neuroprotection. That mechanism may play an important role in ADO’s reported efficacy in suppressing CWNA-induced excitotoxicity. Stimulating A1 ARs is widely accepted to inhibit the release of ACh and other excitatory neurotransmitters including glutamate [38,39,44-48]. (TNO) first investigated CPA’s effect on central ACh levels after CWNA poisoning in 2002 [37]. In that study, striatal ACh levels were measured via microdialysis after rats were poisoned with sarin or VX. The rats were treated with CPA at 2mg/kg either 2 minutes before or 1 minute after nerve agent exposure. Rats that did not receive CPA experienced a significant elevation in ACh after exposure to both sarin and VX. Treatment of CPA before or after sarin exposure suppressed the rise in ACh levels and minimized the cholinergic symptoms. However, CPA did not prevent the rise in ACh levels after VX; it only postponed the onset. Combined with AChE data that was also collected (discussed below), the results from that study led Bueters et al. [37] to hypothesize that CPA’s effect on ACh levels could be attributed to a cardiovascular-related delay in the nerve agent’s delivery to the brain, and not to pre-synaptic inhibition. Although CPA’s effect on ACh accumulation was ambiguous in the Bueters et al. [37] study, we believed that there was sufficient evidence to re-examine the pre-synaptic neuroprotective mechanism.
We hypothesized that CPA at high doses could in fact reduce extracellular ACh after CWNA exposure given its proven ability to block neurotransmitter release and capacity to terminate hyperactive neuronal activity long after AChE inhibition. To test this hypothesis, we used an established in vivo rat microdialysis methodology [49] to measure ACh levels after exposure to a seizure-inducing dose of soman. Rather than administering CPA before or soon after CWNA exposure, we administered treatment 20 minutes after the onset of seizure (approximately 27 minutes after soman exposure). This approach ensured that at the time of CPA treatment, virtually all central AChE would be inhibited, and that there would be a significant elevation in extracellular ACh levels. Any reduction in ACh levels would suggest a pre-synaptic mechanism rather than a cardiovascular-related reduction in nerve agent reaching the brain.
The results from this experiment demonstrated that CPA delivered intraperitoneally (IP) 20 minutes after the onset of soman-induced seizure reduced extracellular levels of ACh in the striatum [50]. Soon after soman exposure, all animals experienced a dramatic peak of ACh levels. That increase corresponded to hyperactive neuronal activity and the onset of seizure. Rats not exposed to soman (exposure control) did not display such elevations in ACh activity. Two hours after soman exposure, rats receiving saline treatment (treatment control group) experienced a severe increase in ACh, 525±44% (average, ±standard deviation) above baseline (Figure 1). Rats that received CPA treatment displayed a lesser rise in ACh: rats receiving 30mg/kg only experienced a 342±73% rise in ACh, and CPA at 60 mg/kg reduced ACh even further to only 273±48% rise. All rats exposed to soman displayed a gradual decline in elevated extracellular ACh for the duration of the experiment; however, the rate of decline was greater for CPAtreated animals. Whereas untreated soman-exposed animals still had elevated ACh levels 250% above baseline after 4 hours, the ACh in animals receiving CPA (60mg/kg) was reduced to approximately baseline values.
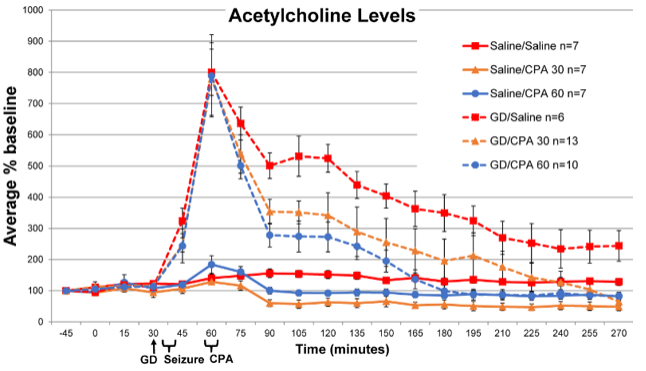
Figure 1: The effect of CPA treatment on striatal ACh concentrations after soman (GD) exposure [50]. Rats were exposed to a seizure-inducing dose of soman (180µg/kg, SC) or saline (0.5 ml/kg, SC), and then treated with CPA (30 or 60 mg/kg, IP) or saline 20 minutes after seizure onset. All soman-exposed animals experienced a dramatic peak in ACh soon after seizure onset. Over the course of the experiment, CPA treatment significantly reduced ACh concentrations in a dose-dependent manner. By the experiment’s endpoint, CPA at 30 and 60mg/kg lowered the ACh concentration to baseline levels.
Reducing ACh levels is an important therapeutic objective because elevated ACh levels lead to excitotoxic neuronal activity which ultimately causes severe brain damage. The decrease in ACh levels in CPA-treated animals corresponded with a reduction in EEG power and seizure termination. Whereas soman-exposed salinetreated rats continued to seize throughout the entire observation period, seizure was terminated in 8 of the 11 animals receiving CPA at 30mg/kg, and 8 of the 9 animals receiving CPA at 60mg/kg. CPA’s capacity to reduce ACh levels and terminate seizure activity also led to the suppression of neuropathology. Soman-exposed animals that did not receive CPA treatment developed severe neuropathology. Pathology scores for the cerebral cortex, piriform cortex, amygdala, hippocampus, thalamus, striatum were totaled and averaged (±standard deviation) 23.4±0.4 (0=normal, 24=most severe). In contrast, animals receiving CPA at 30 mg/kg averaged 10.3±1.6, and animals receiving 60 mg/kg experienced even greater neuroprotection with an average score of 7.3±1.06. These results suggest that CPA’s mechanism of action includes a profound reduction in neurotransmitter levels.
The Effect of Adenosine Agonists on AChE Activity
While CPA is believed to achieve its neuroprotective effect primarily by inhibiting excitotoxic hyper-active neurons via preand post-synaptic effects, other mechanisms may also contribute to protection. That is, ADO agonists such as CPA may directly interact with AChE or the nerve agent to produce a protective shielding effect. Bueters et al. [38] conducted an in vitro and in vivo experiment to investigate this phenomenon [38]. . Using brain homogenates and heparinized rat blood, CPA at 2µM did not appear to directly affect sarin’s inhibition of AChE activity. CPA at 2 mg/kg administered 1 minute after sarin in a living rat slightly delayed the inhibition of AChE, but all AChE activity was eventually eliminated, similar to untreated. In contrast, CPA at 2 mg/kg administered 2 minutes before sarin provided partial protection for some rats and maintained 34% of brain AChE activity on average. Because CPA did not affect AChE activity in vitro, a Kinetic-Pharmaco Dynamic (K-PD) model was created based on the premise that AChE activity was only affected by sarin, and that CPA directly affected the distribution of that sarin. From those data, Bueters et al. [38] concluded that CPA given 2 minutes before nerve agent protected AChE via cardiovascular depressive mechanisms.
However, the development and interpretation of a model based on those in vitro data may be incomplete. It is possible that the assay methodology and experimental procedure did not capture CPA’s dynamic interaction with AChE in a living organism. That is, CPA may temporarily occupy AChE’s active site with a weak bond and thereby provide temporal AChE protection from the nerve agent. In the case of Bueters’ in vitro experiment, CPA may not have been tested at a sufficient concentration and, thus, biased the competition for AChE’s active site toward the more affinitive sarin. In addition, the radiometric methodology itself may not detect CPA’s effect on AChE activity because CPA’s bond is too weak to survive the assay processes. Lastly, the sarin distribution in the K-PD model was controlled by a parameter dependent on the CPA dose and not cardiovascular measures, thus, assuming a direct CPA-cardiovascular relationship. Perhaps future models could better disentangle the observed protective mechanism by explicitly taking into account cardiovascular measures such as heart rate and blood pressure.
Although the mechanism of protection remains to be elucidated, Bueters et al. demonstrated that CPA partially protected AChE from nerve agent [38]. To further investigate CPA’s protective capacity and its effect on AChE activity, we completed a series of in vivo experiments that treated soman-exposed rats to high doses of CPA (60mg/kg, IP) at 1 or 15 minutes after exposure [51]. AChE activity in blood, peripheral tissues, and brain was analyzed from samples taken 60 minutes after soman exposure. The results indicated that immediate CPA treatment protected both peripheral and central AChE from soman-induced inhibition (Figure 2). Whereas soman significantly reduced central AChE activity to 6.2% of baseline in rats not receiving CPA (relative to baseline; rats exposed to saline and treated with saline), CPA-treated rats maintained 82.9% AChE activity. Treatment 15 min after soman exposure did not protect AChE activity, and the AChE activity was reduced to 8.6% of baseline. Even though AChE was not protected in the 15-minute treatment group, all cholinergic toxic signs (seizure and convulsion) were terminated by CPA. AChE activity in saline-exposed CPA-treated animals displayed a minor and statistically insignificant reduction in central AChE to 98.4% of baseline.

Figure 2: The effect of CPA treatment on AChE activity [51]. Rats were exposed to a 112 µg/kg, SC dose of soman and then treated with CPA at 60mg/kg (IP) 1 or 15 minutes later. Forty-five minutes after treatment, rats were euthanized and central (central cortex, striatum, hippocampus, midbrain, cerebellum, brain stem, spinal cord) and peripheral (heart and diaphragm) tissues were collected, processed, and assayed for AChE activity. Relative to rats not receiving soman (i.e., saline control), CPA treatment 1 minute after soman preserved 83.9% central AChE activity and 79.1% peripheral AChE activity after soman. Delaying CPA treatment 15 minutes after soman did not preserve AChE activity. Those rats experienced similar levels of AChE inhibition as rats not receiving treatment (i.e., soman control). Black bar=brain AChE activity and hashed bar=peripheral tissue AChE activity.
This experiment demonstrated that CPA given 1 minute after exposure has a profound protective effect on AChE after soman exposure. Since this protective capacity appears to end within 15 min after exposure, it is critical that CPA be administered as quickly as possible if AChE activity is to be preserved. However, these results also suggest that CPA at 15 minutes would still provide neuroprotection via central inhibitory effects since seizures and convulsions were consistently terminated. Further in silico, in vitro, and in vivo studies are needed to truly understand CPA’s additional protective mechanism and separate CPA’s cardiovascular effects from its inhibitory effects and possible direct interactions with AChE. If CPA is proven to be a temporary inhibitor of AChE, the case for transitioning ADO-based therapies would be strengthened. A medical countermeasure that utilizes both neuronal (neurotransmitter) and AChE (enzyme) protective mechanisms would greatly enhance the efficacy of treatment after CWNA exposure and increase survivability.
Adenosine’s Hypothermic Effect
Systemically and ICV injected A1 AR agonists significantly reduce body temperatures [52,53]. The mechanism of ADO-induced hypothermia and hypo-metabolism has been shown to be centrally mediated with strong involvement of neurons in the nucleus of the solitary tract [54] and the pre-optic area [55]. The effect of ADO on temperature is dose-dependent [56]. In our experiments, a 700 µg dose of CPA ICV consistently reduced rat body temperatures by 4.7°C after 5 hours [40]. Because hypothermia has been shown to be neuroprotective in various brain pathologies such as epilepsy and ischemia [57-59], we hypothesized that hypothermia may play a role in the neuroprotection against CWNAs as well. To test that hypothesis, we compared CPA’s efficacy in a soman-induced seizure rat model when hypothermia was allowed to develop (hypothermic) with rats that receiving thermal support to maintain normal body temperatures (normothermic) between 36-38°C [60]. After exposed to soman and treated with CPA (700µg, ICV) 1 minute later, the rats were observed for 9 hours. Survivors were then perfused and examined for histology. Throughout the observation period, the average body temperature for normothermic saline- and somanexposed rats was 37.7±0.5°C and 37.5±0.3°C, respectively (Figure 3). Rats not receiving thermal support experienced a progressive reduction in body temperature. The average (±standard deviation) minimum temperatures for the hypothermic groups exposed to saline and soman were 31.5±1.67°C and 31.4±2.53°C, respectively. No hypo or normothermic rat experienced seizure after receiving CPA treatment. Furthermore, histological analysis showed that CPA protected the brains of both hypothermic and normothermic animals exposed to soman.
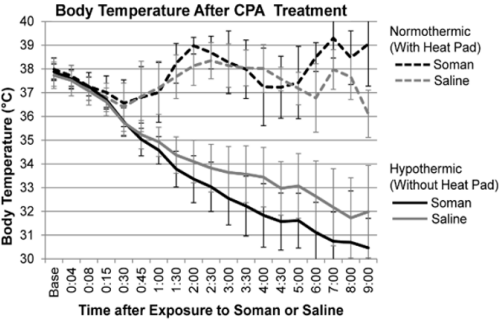
Figure 3: The average body temperatures over the 9-hour observation period following saline (gray line) or soman exposure (black line) and CPA treatment [60]. Rats were pretreated with HI-6 (125mg/kg, IP) 30 minutes before exposed to saline (0.5 ml/kg, SC) or soman (180µg/kg, SC), and then received CPA treatment (700µg, ICV) one minute later. Whereas the body temperature of animals receiving thermal support (normothermic group) was actively maintained near baseline, hypothermic animals experienced a significant reduction throughout the 9-hour observation period.
Although thermal mechanisms did not appear to affect CPA’s neuroprotection in this immediate treatment model, they may have a greater impact when treatment is delayed. In that model, sustained seizure activity initiates a variety of neurotoxic and inflammatory mechanisms that hypothermia could help to suppress. While the neuroprotective effects were similar, the data suggest that body temperature may play a role in promoting survival after soman exposure. Of the 10 hypothermic rats exposed to soman, 7 survived till the study’s endpoint of 9 hours post-exposure. In comparison, only 4 of the 12 normothermic rats exposed to soman survived. Although duration of survival was not significant according to the Kaplan-Meier analysis (p=0.20), there was an obvious trend that decreasing the body temperature may partially mitigate soman’s lethal effects. Future experiments are needed to increase the population size, test different treatment regimens, and extend the observation period to better detect the effect of body temperature on survivability and neuropathology.
Conclusion
CWNAs pose a significant threat to civilian and military personnel. While many therapeutics are able to manage the peripheral cholinergic crisis, they have limited neuroprotective efficacy in the CNS, especially when treatment is delayed and seizure activity becomes sustained. A1 AR agonism may provide the necessary inhibition for preventing or terminating CWNA-induced seizure activity and neuropathology. TNO first investigated the use of ADO as a neuroprotectant against CWNAs and demonstrated partial anti-seizure efficacy in 1998. However, after a series of subsequent studies in the early 2000s, TNO concluded that ADO’s neuroprotective mechanism was an artifact of reduced cardiovascular output and discontinued that research. We renewed the concept of central A1 AR agonism as a CWNA neuroprotectant in 2012.
We conducted novel studies that isolated the effects of central A1 AR stimulation and tested relatively high systemic doses of the A1 AR agonist CPA. The results from our experiments demonstrated that both immediate and delayed treatment with CPA inhibits soman-induced excitotoxic neuronal activity and brain damage. Moreover, those effects were attainable via either direct brain or systemic injections without affecting survivability. That was an important step to clinical relevancy as it was feared that ADO’s depression of cardiovascular output would exacerbate CWNA’s lethality. Additional studies are needed to optimize the exploitation of ADO’s positive central effects while minimizing undesirable cardiovascular side effects. While the data suggested that CPA’s efficacy stemmed from pre- and post-synaptic neuronal mechanisms, other factors beyond the suppression of central neuronal activity could be involved. Data showed that CPA may directly affect soman’s ability to bind to and inhibit AChE early. Protection of the AChE greatly improves the likelihood of a positive outcome. In addition, the hypothermia that develops after ADO treatment may also enhance neuroprotection and survivability after exposure to CWNAs. The comprehensive therapeutic efficacy of CPA that was observed motivates further research into ADObased treatment strategies against CWNA exposure.
Acknowledgement
This research was supported by the Major Command InHouse Laboratory Independent Research program from Director for Basic Research, Office of the Assistant Secretary of the Army for Acquisition, Logistics and Technology, and by an Interagency Agreement (IAA #AOD14018-001-00000) between NIH/NIAID and the USAMRICD.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mcdonough JH, Shih TM (1997) Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neuroscience & Biobehavioral Reviews 21(5): 559-579.
- Shih TM (1982) Time course effects of soman on acetylcholine and choline levels in six discrete areas of the rat brain. Psychopharmacology 78(2): 170-175.
- Donnell JC, Mc Donough JH, Shih TM (2011) In vivo microdialysis and electroencephalographic activity in freely moving guinea pigs exposed to organophosphorus nerve agents sarin and VX: analysis of acetylcholine and glutamate. Archives of Toxicology 85(12): 1607-1616.
- Donnell JC, Acon Chen C, Mc Donough JH, Shih TM (2010) Comparison of extracellular striatal acetylcholine and brain seizure activity following acute exposure to the nerve agents cyclosarin and tabun in freely moving guinea pigs. Toxicology Mechanisms and Methods 20(9): 600-608.
- Wade JV, Samsosn FE, Nelson SR, Pazdernik TL (1987) Changes in extracellular amino acids during soman- and kainic acid-induced seizures. Journal of Neurochemistry 49(2): 645-650.
- El Etri MM, Nickell WT, Ennis M, Skau KA, Shipley MT (1992) Brain norepinephrine reductions in soman-intoxicated rats: association with convulsions and AChE inhibition, time course and relation to other monoamines. Experimental Neurology 118(2): 153-163.
- Lallement G, Carpentier P, Collet A, Pernot Marino I, Baubichon D, et al. (1991) Effects of soman-induced seizures on different extracellular amino acid levels and on glutamate uptake in rat hippocampus. Brain Research 563(1): 234-240.
- Lallement G, Denoyer M, Collet A, Pernot Marino I, Baubichon D, et al. (1992) Changes in hippocampal acetylcholine and glutamate extracellular levels during soman-induced seizures: influence of septal cholinoceptive cells. Neuroscience Letters 139(1): 104-107.
- Fosbraey P, Wetherell JR, French MC (1990) Neurotransmitter changes in guinea-pig brain regions following soman intoxication. Journal of Neurochemistry 54(1): 72-79.
- Shih TM, Koviak TA, Capacio BR (1991) Anticonvulsants for poisoning by the organophosphorus compound soman: pharmacological mechanisms. Neuroscience and Biobehavioral Reviews 15(3): 349-362.
- Marrs TC (2004) The role of diazepam in the treatment of nerve agent poisoning in a civilian population. Toxicological Reviews 23(3): 145-157.
- Mc Donough JH, Mc Monagle JD, Shih TM (2010) Time-dependent reduction in the anticonvulsant effectiveness of diazepam against soman-induced seizures in guinea pigs. Drug and Chemical Toxicology 33(3): 279-283.
- Shih TM, Maxwell DM, Koplovitz I, Kan RK, Mc Donough JH (2010) Reactivation of acetylcholinesterase activity and its therapeutic benefits in nerve agent intoxication. In Weissman BA, Raveh L (Eds), The Neurochemical Consequences of Organophosphate Poisoning in the CNS, Transworld Research Network. Kerala, India, pp. 111-133.
- Marrs TC, Rice P, Vale JA (2006) The role of oximes in the treatment of nerve agent poisoning in civilian casualties. Toxicological Reviews 25(4): 297-323.
- Mc Donough JH, Dochterman LW, Smith CD, Shih TM (1995) Protection against nerve agent-induced neuropathology, but not cardiac pathology, is associated with the anticonvulsant action of drug treatment. Neurotoxicology 16(1): 123-132.
- Ribeiro JA, Sebastiao AM, De Mendonca A (2002) Adenosine receptors in the nervous system: pathophysiological implications. Progress in Neurobiology 68(6): 377-392.
- Cunha RA (2005) Neuroprotection by adenosine in the brain: From A1 receptor activation to A2A receptor blockade. Purinergic Signalling 1(2): 111-134.
- Lynge J, Hellsten Y (2000) Distribution of adenosine A1, A2A and A2B receptors in human skeletal muscle. Acta Physiologica Scandinavica 169(4): 283-290.
- Svenningsson P, Hall H, Sedvall G, Fredholm BB (1997) Distribution of adenosine receptors in the postmortem human brain: an extended autoradiographic study. Synapse 27(4): 322-335.
- Van Helden HP, Groen B, Moor E, Westerink BH, Bruijnzeel PL (1998) New generic approach to the treatment of organophosphate poisoning: adenosine receptor mediated inhibition of ACh-release. Drug and Chemical Toxicology 21(1): 171-181.
- St Hilaire C, Carroll SH, Chen H, Ravid K (2009) Mechanisms of induction of adenosine receptor genes and its functional significance. Journal of Cellular Physiology 218(1): 35-44.
- Malva JO, Silva AP, Cunha RA (2003) Presynaptic modulation controlling neuronal excitability and epileptogenesis: role of kainate, adenosine and neuropeptide Y receptors. Neurochemical Research 28(10): 1501-1515.
- Bjorness TE, Greene RW (2009) Adenosine and sleep. Current Neuropharmacology 7(3): 238-245.
- Dixon AK, Gubitz AK, Sirinathsinghji DJ, Richardson PJ, Freeman TC (1996) Tissue distribution of adenosine receptor mRNAs in the rat. British Journal of Pharmacology 118(6): 1461-1468.
- Schindler CW, Karcz Kubicha M, Thorndike EB, Muller CE, Tella SR, et al. (2005) Role of central and peripheral adenosine receptors in the cardiovascular responses to intraperitoneal injections of adenosine A1 and A2A subtype receptor agonists. British Journal of Pharmacology 144(5): 642-650.
- Shen HY, Chen JF (2009) Adenosine A2A receptors in psychopharmacology: modulators of behavior, mood and cognition. Current Neuropharmacology 7(3): 195-206.
- Schiffmann SN, Fisone G, Moresco R, Cunha RA, Ferre S (2007) Adenosine A2A receptors and basal ganglia physiology. Progress in Neurobiology 83(5): 277-292.
- Rosin DL, Hettinger BD, Lee A, Linden J (2003) Anatomy of adenosine A2A receptors in brain: morphological substrates for integration of striatal function. Neurology 61(6): 12-18.
- Tabrizchi R, Bedi S (2001) Pharmacology of adenosine receptors in the vasculature. Pharmacology & Therapeutics 91(2): 133-147.
- Basheer R, Strecker RE, Thakkar MM, Mc Carley RW (2004) Adenosine and sleep-wake regulation. Progress in Neurobiology 73(6): 379-396.
- Schubert P, Ogata T, Marchini C, Ferroni S, Rudolphi K (1997) Protective mechanisms of adenosine in neurons and glial cells. Annals of the New York Academy of Sciences 825: 1-10.
- Wardas J (2002) Neuroprotective role of adenosine in the CNS. Polish journal of pharmacology 54(4): 313-326.
- Boison D (2012) Adenosine augmentation therapy. In Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV (Eds), Jasper’s Basic Mechanisms of the Epilepsies, (4th edn.). Bethesda, MD, pp. 1707-1723.
- Gouder N, Fritschy JM, Boison D (2003) Seizure suppression by adenosine A1 receptor activation in a mouse model of pharmacoresistant epilepsy. Epilepsia 44(7): 877-885.
- Huber A, Guttinger M, Mohler H, Boison D (2002) Seizure suppression by adenosine A(2A) receptor activation in a rat model of audiogenic brainstem epilepsy. Neuroscience Letters 329(3): 289-292.
- Young D, Dragunow M (1994) Status epilepticus may be caused by loss of adenosine anticonvulsant mechanisms. Neuroscience 58(2): 245-261.
- Bueters TJ, Groen B, Danhof M, Ijzerman AP, Van Helden HP (2002) Therapeutic efficacy of the adenosine A1 receptor agonist N6- cyclopentyladenosine (CPA) against organophosphate intoxication. Archives of Toxicology 76(11): 650-656.
- Bueters TJ, Joosen MJ, Van Helden HP, Ijzerman AP, Danhof M (2003) Adenosine A1 receptor agonist N6-cyclopentyladenosine affects the inactivation of acetylcholinesterase in blood and brain by sarin. The Journal of Pharmacology and Experimental Therapeutics 304(3): 1307- 1313.
- Joosen MJ, Bueters TJ, van Helden HP (2004) Cardiovascular effects of the adenosine A1 receptor agonist N6-cyclopentyladenosine (CPA) decisive for its therapeutic efficacy in sarin poisoning. Archives of Toxicology 78(1): 34-39.
- Thomas TP, Shih TM (2014) Stimulation of central A1 adenosine receptors suppresses seizure and neuropathology in a soman nerve agent seizure rat model. Toxicology Mechanisms and Methods 24(6): 385-395.
- Thomas TP, Shih TM (2014) The neuroprotective benefits of central adenosine receptor stimulation in a soman nerve agent rat model. US Army Medical Research Institute of Chemical Defense, Aberdeen Proving Ground.
- Thomas TP, Shih TM (2015) Evaluating the anti-seizure efficacy of novel adenosine treatment regimens in a soman rat model. US Army Medical Research Institute of Chemical Defense, Aberdeen Proving Ground.
- Evoniuk G, von Borstel RW, Wurtman RJ (1987) Antagonism of the cardiovascular effects of adenosine by caffeine or 8-(p-sulfophenyl) theophylline. Journal of Pharmacology and Experimental Therapeutics 240(2): 428-432.
- Fontanez DE, Porter JT (2006) Adenosine A(1) receptors decrease thalamic excitation of inhibitory and excitatory neurons in the barrel cortex. Neuroscience 137(4): 1177-1184.
- Scholz KP, Miller RJ (1996) Presynaptic inhibition at excitatory hippocampal synapses: development and role of presynaptic Ca2+ channels. Journal of Neurophysiology 76(1): 39-46.
- Dunwiddie TV, Masino SA (2001) The role and regulation of adenosine in the central nervous system. Annual Review of Neuroscience 24: 31-55.
- Haas HL, Selbach O (2000) Functions of neuronal adenosine receptors. Naunyn-Schmiedeberg’s Archives of Pharmacology 362(4): 375-381.
- Prince DA, Stevens CF (1992) Adenosine decreases neurotransmitter release at central synapses. Proc National Academy of Science USA 89(18): 8586-8590.
- Donnell JC, McDonough JH, Shih TM (2011) In vivo microdialysis and electroencephalographic activity in freely moving guinea pigs exposed to organophosphorus nerve agents sarin and VX: analysis of acetylcholine and glutamate. Archives of Toxicology 85(12): 1607-1616.
- Thomas TP, Shih TM (2015) Acute or delayed treatment with an adenosine agonist provides neuroprotection in a soman seizure nerve agent rat model. In 2015 Chemical and Biological Defense Science and Technology (CBD S&T) Conference, St Louis.
- Thomas TP, Shih TM (2015) Acute adenosine A1 agonist N6- cyclopentoadenosine (CPA) treatment protects acetylcholinesterase after exposure to soman nerve agent in a rat model In Society of Toxicology 54th Annual Meeting, San Diego.
- Zarrindast MR, Heidari MR (1993) Involvement of adenosine receptors in mouse thermoregulation. Journal of Psychopharmacology 7(4): 365- 370.
- Coupar IM, Tran BL (2002) Effects of adenosine agonists on consumptive behaviour and body temperature. Journal of Pharmacy and Pharmacology 54(2): 289-294.
- Tupone D, Madden CJ, Morrison SF (2013) Central activation of the A1 adenosine receptor (A1AR) induces a hypothermic, torpor-like state in the rat. Journal of Neuroscience 33(36): 14512-14525.
- Ticho SR, Radulovacki M (1991) Role of adenosine in sleep and temperature regulation in the preoptic area of rats. Pharmacology, Biochemistry and Behavior 40(1): 33-40.
- Anderson R, Sheehan MJ, Strong P (1994) Characterization of the adenosine receptors mediating hypothermia in the conscious mouse. British Journal of Pharmacology 113(4): 1386-1390.
- Niquet J, Gezalian M, Baldwin R, Wasterlain CG (2015) Neuroprotective effects of deep hypothermia in refractory status epilepticus. Annals of Clinical and Translational Neurology 2(12): 1105-1115.
- Cilio MR, Ferriero DM (2010) Synergistic neuroprotective therapies with hypothermia. Seminars in Fetal & Neonatal Medicine 15(5): 293-298.
- Yenari MA, Hemmen TM (2010) Therapeutic hypothermia for brain ischemia: where have we come and where do we go? Stroke 41(10): 72-74.
- Thomas TP, Shih TM (2015) The development of a neuroprotective treatment for nerve agent poisoning using central A1 adenosine receptor agonists. In Program of the 9th Annual NIH Countermeasures Against Chemical Threats (CounterACT) Network Research Symposium, New York, USA.
