 Case Report
Case ReportAbstract
Introduction: Wiskott-Aldrich syndrome (WAS) is an X-linked recessive immunodeficiency caused by WASP mutations. Identification of novel WASP mutations may help understand uncovered pathogenesis. We here noticed a rare case with a novel WASP mutation, which was also detected in his mother and grandmother. This article aimed to detect the genetic features of a WAS family and determine the source of the completely new mutation.
Case Presentation: This Chinese male infant was admitted to our department at postnatal day 7. He was found thrombocytopenia for 4 days. The family history documented that his two uncles both died in infancy due to repeated infections; his aunt had experienced 2 pregnancies, and both resulted in unexplained abortion. After 18-day treatment, his parents signed the request to discharge. The infant finally died at 9 months old. A novel heterozygous mutation in the exon 9 was found on his X chromosome, that c.928C>T resulting in amino acid changes p.Q310X. This mutation was revealed to be derived from his grandmother to his mother.
Conclusion: In conclusion, a novel mutation (c.928C>T) was found in our male infant patient, as well as his mother and grandmother. The patient died from uncontrollable WAS symptoms, as the two boys of his grandmother did. Early detection and diagnosis of new mutation may reduce complications and increase the life expectancy of WAS patients. Prenatal DNA sequencing could be recommended to avoid birth of WAS for male fetuses.
Keywords: Wiskott-Aldrich syndrome; c.928C>T; WASP; X-linked recessive immunodeficiency; Sequencing
Introduction
Wiskott-Aldrich syndrome (WAS) is a sort of X-linked rare disorder which dominantly affects male infants. The incidence of WAS is around 3-10 per million babies. It was first reported by Wiskott in 1937, and then in 1954 Aldrich et al. reported that in 6 generations of a family, 16 of 40 males, but no females, died of a disease like that Wiskott’s report, and they proposed an X-linked mode of inheritance [1]. Clinically, this recessive immunodeficiency is characterized by thrombocytopenia, eczema, recurrent infection, bloody diarrhea, chronic diarrhea, melena, inflammatory bowel disease, autoimmune disease and malignant tumors. WAS usually leads to death before 10 years old. Besides these phenotypes with highest severities, milder forms of WA include: X-linked thrombocytopenia (XLT), which is absent of or with mild eczema/ immunodeficiency, and X-linked neutropenia (XLN). A wide spectrum of clinical phenotypes mentioned above are commonly decided by types of WAS related mutations.
Genetically, the WAS related gene locates on Xp11.22–23 (the short arm of the X chromosome), which spans about 9 kbps. It has 12 exons and encodes a protein, namely Wiskott-Aldrich syndrome protein (WASP), with 502 amino acids. WASP is majorly expressed by non-erythroid hematopoietic cells [2,3]. This protein is involved in actin polymerization, signaling pathways, and cytoskeletal rearrangement; and it is crucial for the monocytes and macrophages migration. WASP deficiency (either complete or partial) may cause malfunctioning of tissue macrophages, neutropenia, and small platelet sizes, resulting in chronic and repeated infections [4-7]. Classic WAS, XLT and XLN are caused by different WASP mutations, each having a distinct pattern of clinical symptoms and disease severity [8]. The classic WAS patients completely lose the expression of WASP protein; XLT is caused by a missense mutation that results in insufficient or incomplete expression WASP [9-11]; XLN is caused by constitutive activation of WASP protein expression due to functional acquired mutations [12-14]. So far, over 300 types of mutations, including deletions, insertions, and splice sites, have been recorded, which cover all 12 exons. We here noticed a rare Chinese case with a novel WASP mutation, which was also detected in his mother and grandmother. This article aimed to detect the genetic features of this family and determine the source of this completely new mutation.
Case Presentation
This Chinese male infant was admitted to our department at postnatal day 7. He had experienced 39 weeks + 4 days of pregnancy and was born by Caesarean section. Before admission, he had suffered from thrombocytopenia for 4 days. The admission examination was performed, and the infant was found conscious and with good response. No obvious rash and bleeding points were noticed. Also, cardiopulmonary and abdomen examination showed no obvious abnormalities. A previous examination reported the right lower pneumonia according to the chest radiograph. Results of blood tests before admission were as follow: white blood cell (WBC) 9.58×10 ^9/L, hemoglobin (HGB) 166 g/L, platelet (PLT) 53×10 ^9 /L, neutrophil (N) 54.4%, lymphocyte (L) 25.2%. The family history documented that the infant’s grandmother had four children: his two uncles both died in infancy due to repeated infections; his aunt had experienced 2 pregnancies, and both resulted in unexplained abortion. His mother was physically healthy. The blood tests after admission were listed in Table 1.
The immune function examination showed increased IgG (16.80g/L), IgA (0.26g/L) and C3 (1.19g/L) levels. The blood lymphocyte subsets tests exhibited a down- regulated CD8+ T cell percentage (5.25%), CD8+ T cell number (107 /μl), CD19 + cell percentage (10.14%), and CD19 + number (207/μl ), as well as up-regulated Th/Ts level (10.4) and CD4 + T percentage (54.76%). Enterococcus faecium infection was detected in the urine culture, and the enterococcus faecium was sensitive to vancomycin. Left renal pelvis filling was observed from the result of double renal ureteral imaging. Other indexes we determined were all in the normal range, including plate-type megakaryocyte level (8%) sampled from bone marrow, the cerebrospinal fluid examinations, blood amino acid levels, and urinary organic acid examination.
The child ran a fever (38 °C) for 1 day during hospitalization. The stool was once noticed with a little bloodshot. After admission, the mutation detection was performed at the first time. Besides, the infant was given a 12-day antibiotic therapy as the following process: cefoperazone sulbactam sodium (0.17g/8h, for 8 days, April 12 to 20), meropenem (0.06g/8h for 5 days, April 20 to 25), and vancomycin (0.05g/8 h for 7 days, April 18 to 25). Simultaneously, the gamma globulin support and amino acid milk powder feeding were given. After above treatments, the clinical symptoms improved to some extent, but there still existed thrombocytopenia and a high CRP level (Table 1). Afterwards, his parents signed the request to discharge, and we followed up several times after discharge. It was reported that the infant displayed repeated fever, diarrhea, bloody stool and skin ecchymosis. He received further intermittent gamma globulin treatment four times, but finally died at 9 months old.
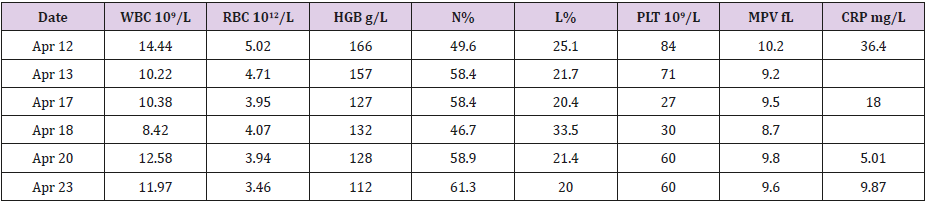
Table 1: Blood test results after admission.
Given the above clinical phenotypes, the family history, the intracranial infection state, and that the male patient died in infancy, it was conceivable to assume an X- linked recessive heredity. Besides, the patient may also have primary immunodeficiency disease with thrombocytopenia. To reveal the genetic cause of WAS in this patient, we performed Next Generation Sequencing to probe all exons of WAS. The original short sequence was obtained by the Illumina Hi Seq2500 platform using standardized operating procedure. The joints and low-quality data were removed, and the short sequence was mapped to the corresponding position of the human genome data using the SOA Paligner software. SOAPsnp was used to find the genotype of the locus in the target area. SNPs with low quality values (mass value <=20) and low coverage (depth <=10) were screened out.
The GATK software was used to find out the sequence insertion/ deletion (InDel) information. After annotation of SNPs using CCDS, the following parameters were identified: mRNA sites, amino acid changes, coding regions at which mutations occur, sequence changes, SNP function (missense mutation/nonsense mutation/ variable cleavage site) and In Del function (amino acid insertion/ amino acid deletion/frameshift mutation). The WASP gene sequencing results were as follow. A heterozygous mutation in the exon 9 was found on the X chromosome, that c.928C>T resulting in amino acid changes p.Q310X (glutamine>termination) (Figure 1A). For reference, the blood sample of the mother was detected, and as expected, the same nonsense mutation was found at the same site of the WAS gene on one of her alleles (Figure 1B). In comparison, his father exhibited a normal genotype (Figure 1C).
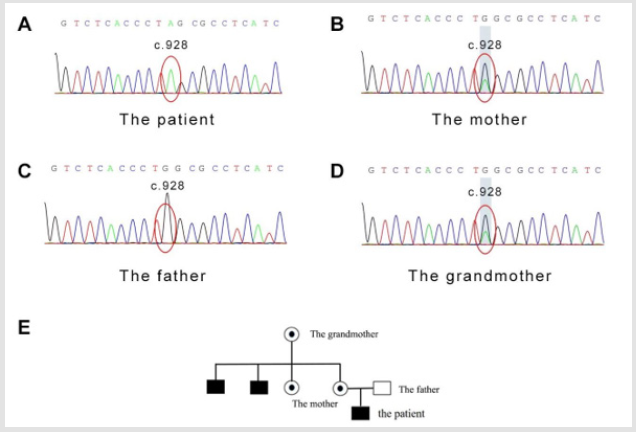
Figure 1: The WASP c.928C>T mutation detection in the patient’s family and the mutation pedigree. (A) c.928C>T observed in the infant male patient. (B) c.928C>T observed in the patient’s mother. (C) The patient’s father showed a normal wild type. (D) c.928C>T observed in the patient’s grandmother. (E) Diagrammatic WASP c.928C>T mutation pedigree.
Considering two babies of the grandmother also died soon after birth, her WAS gene was also sequenced. Again, the same mutation at the identical site was probed in the grandmother. Collectively, we drew a pedigree of the WAS family based on their history. As Figure 1E shown. the mutation gene of the child was derived from the mother and further from grandmother. To date, this mutation site has not been reported in any articles or The Human Gene Mutation Database. Theoretically, this termination mutation, the effect on protein function should be huge and this novel mutation type is worth documentation and further exploration.
Discussion
WASP protein is a key regulator of actin polymerization in hematopoietic cells, with functions involved in cell signaling and cell locomotion, immune synapse formation, and apoptosis [15]. WASP domains can be regulated by different factors like GTPase, Cdc42, ARP2/3, p47nck, and Fyn, etc. [16]. To date, over 300 unique mutations in WASp have been observed, spanning all 12 exons, with missense, nonsense, splice site mutations, insertions, deletions, and rearrangements. Among these polymorphisms, missense mutations are the predominant form, in exons 1 to 4. The most well-defined function disorders are as follow: WAS occurs when WASP is absent, XLT when mutated WASP is expressed and XLN when missense mutations occur in the Cdc42-binding site [15]. WASP is considered as a link between Cdc42 and the actin cytoskeleton, WASP mutations generally cause a sever disruption of actin cytoskeleton accompanied by impaired Cdc42 signaling. Besides CDC42 and ARPC2/3, WASP was known to interact with some import proteins, such as ACTR2, WIPF1, ACTR3, PFN1, so on [17].
Recently, there have been updated understanding of WAS based on rare mutations. For example, early onset recurrent acute hemorrhagic edema and hyperostosis were found as unusual manifestations and associated with p. Glu31Lys mutation [18]. Together, as a crucial role in the complex bioprocess network in hematopoietic cells, it regulates the T Cell Receptor Signaling Pathway, G13 Signaling Pathway, Pathogenic Escherichia coli infection, T-Cell antigen Receptor (TCR) Signaling Pathway, Adhering junction, Endocytosis, CDC42 Pathway, FAK1 Signaling, Cytoskeletal Signaling, and so on; and WASP mutation may impact various pathways and cause life-threating clinical phenotypes [19]. The above WASP functions had been consistently identified in different cell types. Years ago, a study about somatic mosaicism using 497 specific T-cell clones generated from the peripheral blood, reported that 47.1% carried a revertant sequence and all revertant T-cell clones exhibited restoration of WASP expression [20].
For invariant natural killer T (iNKT) cells, WASP is required for their maturation and activation [21]. On the other hand, WASPexpressing cells were noticed present mainly CD56+ NK cells and CD8+ T cells [22]. At the level of amino acid modifications, phosphorylation at Tyr-291 by FYN and HCK, inducing WAS effector activity in promoting actin polymerization and filopodia formation [23]; two phosphorylation sites in the VCA domain of WASP at serine 483 and 484 increase its affinity for the Arp2/3 complex and enhances actin polymerization [24]. As the Phosphosite database shown, most frequently documented modification site of was Y291, a phosphorylation site. Mutation caused WASP protein polymorphism also causes different types of cancers, among which Endometrial cancer ranks first, and then lung cancer. So far, it remains unknown what causes WASP mutations and how WASP is mutated. But the mechanisms would become gradually clear when all variation types were discovered.
In recent years, increasing reports about novel mutations in WASP, as well as their clinical links, have enriched the mutation database of WAS. However, it is not easy to accumulate boundary cases and samples, for this is a rare disease. In 2010, Chinese scholars reported a clustering pattern on exon 1 and five unique mutations (deletion of 45-48 ACCA, IVS 1 (-1) G>C, large deletion of promoter and exon 1 and 2, insertion 1023 C, and 1378 C>T and 1421 T>C, and demonstrated genetic variations in different ethnic groups [25]. Another Chinese team identified a novel mutation in the same year: the first nucleotide in exon 8 (G) had been deleted (769delG), which resulted in two kinds of aberrant mRNA with abnormal splicing and causes frameshift and a stop codon at amino acid 260 [26]. Simultaneously, seven novel mutations (168 C>A, 747-748del T, 793-797del C, 1185 ins C, Dup 1251-1267, 1277 ins A and 1266 C>G; 1267-1269del C) were discovered by another Chinese team [27]. In 2011, another 17 novel sequence variants were observed [28].
In 2012, Korea scholars reported an IVS6+5G>A mutation of WAS in a Korean family [29]. In 2013, an extremely rare case was reported, for that mutations in the X chromosomes typically affect males but this article found a girl with WAS. Although the girl had a mutation (c.397G > A, p.E133K) in only one allele, she carried an extremely skewed X-inactivation pattern and no expression of the WAS protein [30] (another similar case was recorded in Japan in 2015 [31]). Next, Japanese researchers identified a novel mutation of WASP gene: the seventh nucleotide in exon 11 (G) had been deleted (1345delG) in 2013 [32]. Later, c.952-953delCC mutation was discovered in 2014 [33]. Afterwards, deletion mutation (1144delA) at exon 10, and c.410_419del10 in exon 4 were first reported by Chinese teams [34,35]. In 2015, 29 novel mutations were found by a Chinese lab, including 321T>C, 415C>A, 471C>T, 102-105delC, 521 del C, 1330 del A, IVS2-2 a>c, 168 C>A/1412 C> T, exon1-2 del/1412 C>T, and so on [36]. In the same year, 20 novel mutations were detected by another Chinese team [37]; and an interesting and novel mutation was found by Canadian scholars: hemizygous for c.1125_1129delTGGAC mutation [38].
This mutation caused a unique clinical presentation: the patient exhibited a lower gastrointestinal bleed, leukopenia, and thrombocytopenia, but with normal platelet volume. In 2016, it was noticed that a 9-month old boy carried an insertion G in exon 10 of WASP gene, which leaded a premature stop immediately at amino acid 335 (N335X or p. G334GfsX1) and truncated protein [39]. In that year, two unpublished mutations were further revealed: c.778-2A>T and c.1017delT [40]. In 2017 to 2018, the following mutations were discovered: c.1178dupT [41], c.257G>A [42], c. 343 C>T [43], and c.360 + 1G > C [44]. Still, c.928C>T is a mutation with undermined clinical significance. We here for the first time observed a nonsense mutation that c.928C>T, resulting in amino acid changes p.Q310X (glutamine>termination), in the infant patient, his mother and grandmother. This study expanded the mutation pool of WASP and clearly emphasized the severity of the c.928C>T encoding termination mutation.
For treatment, antibiotic therapy assisted with gamma globulin support, as our patient received, is still a necessary strategy for most of WAS cases. Novel strategies have been proposed in the last decade. Stem cell transplantation offers the opportunity of cure for all complications, and there has been a remarkable improvement in survival following this treatment [45]. For example, bone marrow transplantation is an effective strategy for WAS patients. The life expectancy of treated individuals is around 20 years but that without treatment is 3.5 years [46]. Previous reports have claimed excellent long-term survival in Chinese patients who underwent hematopoietic stem cell transplant (HSCT) [47]. In 2010, a study analyzed the long- term outcome of 194 WAS patients who received HSCT in the period 1980- 2009.
The 5-year survival rate was 89.1%. Their observations indicated continuous improvement of outcome after HSCT and implied important implications for novel protocols aiming to obtain full correction of the disease [2]. In 2014, a Chinese WAS boy first tried umbilical cord blood transplantation because no matched sibling donor was available. But the treatment succeeded, and patient was free of any bleeding episodes [35]. Finally, for patients with a poorly matched stem cell donor, gene therapy trials may provide encouraging results with low toxicity [45]. However, the efficiency still urgently needs improvement. A vast combination of approaches was performed in 2010 by Italian scholars, but they found the patient still had severe infections and autoimmune disorders, which suggested re-expression of WASP in T cells is not enough to normalize immune functions fully in WAS patients [48].
In conclusion, a novel mutation (c.928C>T) was found in our male infant patient, as well as his mother and grandmother. The patient died from uncontrollable WAS symptoms, as the two boys of his grandmother did. Early detection and diagnosis of new mutation may reduce complications and increase the life expectancy of WAS patients. Prenatal DNA sequencing could be recommended to avoid birth of WAS for male fetuses.
Acknowledgement
We thank the patient and his family for their participation in this study.
References
- Ochs HD, Filipovich AH, Veys P, Cowan MJ, Kapoor N (2009) Wiskott- Aldrich syndrome: diagnosis, clinical and laboratory manifestations and treatment. Biol Blood Marrow Transplant 15(1 Suppl): 84-90.
- Moratto D, Giliani S, Bonfim C, Mazzolari E, Fischer A (2011) Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott- Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980-2009: an international collaborative study. Blood 118(6): 1675-1684.
- Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, Schulz A, Thrasher AJ (2008) Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood 111(1): 439-445.
- Ma H, Liu L, Xu S, Hu W, Zhang Y, Ma Y (2014) HLA haploidentical hematopoietic stem cell transplantation for Wiskott-Aldrich syndrome: a case report and literatures review. Zhonghua Xue Ye Xue Za Zhi 35(11): 1027-1029.
- Pai SY, LD Notarangelo (2010) Hematopoietic cell transplantation for Wiskott-Aldrich syndrome: advances in biology and future directions for treatment. Immunol Allergy Clin North Am 30(2): 179-194.
- Boztug K, RA Dewey, C Klein (2006) Development of hematopoietic stem cell gene therapy for Wiskott-Aldrich syndrome. Curr Opin Mol Ther 8(5): 390-395.
- Snapper SB FS Rosen (1999) The Wiskott-Aldrich syndrome protein (WASP): roles in signaling and cytoskeletal organization. Annu Rev Immunol 17: 905-929.
- Albert MH, LD Notarangelo, HD Ochs (2011) Clinical spectrum, pathophysiology and treatment of the Wiskott-Aldrich syndrome. Curr Opin Hematol 18(1): 42-48.
- Kim AS Kakalis LT, Abdul-Manan N, Liu GA, Rosen MK (2000) Autoinhibition and activation mechanisms of the Wiskott– Aldrich syndrome protein. Nature 404(6774): 151-158.
- Dupuis Girod S, Medioni J, Haddad E, Quartier P, Cavazzana-Calvo M (2003) Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics 111(5): e622-e627.
- Galy A (2015) Efficacy and safety of gene therapy for Wiskott-Aldrich syndrome. Med Sci (Paris) 31(12): 1066-1069.
- Thrasher AJ (2009) New insights into the biology of Wiskott-Aldrich syndrome (WAS). Hematology Am Soc Hematol Educ Program pp.132- 138.
- Albert MH, Bittner TC, Nonoyama S, Notarangelo LD, Burns’ S (2010) X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood 115(16): 3231-3238.
- Nikolov NP, Shimizu M, Cleland S, Bailey D, Aoki J (2010) Systemic autoimmunity and defective Fas ligand secretion in the absence of the Wiskott-Aldrich syndrome protein. Blood 116(5): 740-747.
- Ochs HD (2009) Mutations of the Wiskott-Aldrich Syndrome Protein affect protein expression and dictate the clinical phenotypes. Immunol Res 44(1-3): 84-88.
- Massaad MJ, N Ramesh, RS Geha (2013) Wiskott-Aldrich syndrome: a comprehensive review. Ann N Y Acad Sci 1285: 26-43.
- Bender M, Stritt S, Nurden P, van Eeuwijk JM, Zieger B (2014) Megakaryocyte-specific Profilin1-deficiency alters microtubule stability and causes a Wiskott-Aldrich syndrome-like platelet defect. Nat Commun 5: 4746.
- Chandrakasan S (2011) Wiskott-Aldrich syndrome presenting with early onset recurrent acute hemorrhagic edema and hyperostosis. Pediatr Blood Cancer 56(7): 1130-1132.
- Blundell MP (2010) The Wiskott-Aldrich syndrome: The actin cytoskeleton and immune cell function. Dis Markers 29(3-4): 157-175.
- Jia D, Gomez TS, Metlagel Z, Umetani J, Otwinowski Z (2010) WASH and WAVE actin regulators of the Wiskott-Aldrich syndrome protein (WASP) family are controlled by analogous structurally related complexes. Proc Natl Acad Sci USA 107(23): 10442-10447.
- Locci M, Elena Draghici, Francesco Marangoni, Marita Bosticardo, Marco Catucci (2009) The Wiskott-Aldrich syndrome protein is required for iNKT cell maturation and function. J Exp Med 206(4): 735-742.
- Xie JW, Zhang ZY, Wu JF, Liu DW, Liu W, Zhao Y (2015) In vivo reversion of an inherited mutation in a Chinese patient with Wiskott-Aldrich syndrome. Hum Immunol 76(6): 406-413.
- Cory GO, Garg R, Cramer R, Ridley AJ (2002) Phosphorylation of tyrosine 291 enhances the ability of WASp to stimulate actin polymerization and filopodium formation. Wiskott- Aldrich Syndrome protein. J Biol Chem 277(47): 45115-45121.
- Cory GO, Cramer R, Blanchoin L, Ridley AJ (2003) Phosphorylation of the WASP-VCA domain increases its affinity for the Arp2/3 complex and enhances actin polymerization by WASP. Mol Cell 11(5): 1229-1239.
- Lee WI, Huang JL, Jaing TH, Wu KH, Chien YH (2001) Clinical aspects and genetic analysis of taiwanese patients with wiskott-Aldrich syndrome protein mutation: the first identification of x- linked thrombocytopenia in the chinese with novel mutations. J Clin Immunol 30(4): 593-601.
- Yu HA, Liu T, Meng W, Hou L (2010) novel WASP gene mutation in a Chinese boy with Wiskott-Aldrich syndrome. Int J Hematol 92(2): 271- 275.
- Zhang ZY, Xiao HQ, Jiang LP, Zhou Y, Zhao Q (2010) Analysis of clinical and molecular characteristics of Wiskott-Aldrich syndrome in 24 patients from 23 unrelated Chinese families. Pediatr Allergy Immunol 21(3): p. 522-32.
- Gulacsy V, Freiberger T, Shcherbina A, Pac M, Chernyshova L (2011) Genetic characteristics of eighty-seven patients with the Wiskott-Aldrich syndrome. Mol Immunol 48(5): 788-792.
- Yoon SH, Cho T, Kim HJ, Kim SY, Ko JH, Baek HS (2012) IVS6+5G>A found in Wiskott-Aldrich syndrome and X- linked thrombocytopenia in a Korean family. Pediatr Blood Cancer 58(2): 297-299.
- Boonyawat B, Dhanraj S, Al Abbas F, Zlateska B, Grunenbaum E (2013) Combined de-novo mutation and non-random X- chromosome inactivation causing Wiskott-Aldrich syndrome in a female with thrombocytopenia. J Clin Immunol 33(7): 1150-1155.
- Takimoto T (2015) Wiskott-Aldrich syndrome in a girl caused by heterozygous WASP mutation and extremely skewed X-chromosome inactivation: a novel association with maternal uniparental isodisomy 6. Neonatology 107(3): 185-190.
- Kawasaki Y (2013) A novel Wiskott-Aldrich syndrome protein mutation in an infant with thrombotic thrombocytopenic purpura. Eur J Haematol 90(2): 164-168.
- Liu N (2014) [Mutation analysis of WASP gene and prenatal diagnosis of Wiskott-Aldrich syndrome]. Zhonghua Er Ke Za Zhi 52(9): 662-666.
- Wu HA, Cheng Hu, Dan Dang, Ying-Jie Guo (2014) Novel WASP Gene Mutation in a Chinese Boy with Wiskott- Aldrich Syndrome. Indian J Hematol Blood Transfus 30(Suppl 1): 353-355.
- Zhu X, Tang B, Zheng C, Liu H, Song K (2014) A novel mutation in Wiskott-Aldrich syndrome and successfully treated with umbilical cord blood transplantation. Blood Cells Mol Dis 53(4): 283-285.
- Li W, Liu D1, Zhang X1, Ding Y2, Zhao X (2015) [Clinical features and genotype analysis of 132 patients with Wiskott-Aldrich syndrome]. Zhonghua Er Ke Za Zhi 53(12): 925-930.
- Liu DW, Zhang ZY, Zhao Q, Jiang LP, Liu W (2015) Wiskott-Aldrich syndrome/X-linked thrombocytopenia in China: Clinical characteristic and genotype-phenotype correlation. Pediatr Blood Cancer 62(9): 1601- 1608.
- Yoonessi L, Randhawa I, Nussbaum E, Saharti S, Do P (2015) Wiskott- Aldrich Syndrome: Description of a New Gene Mutation with Normal Platelet Volume. J Pediatr Hematol Oncol 7(7): 515-518.
- Eghbali M, Sadeghi-Shabestari M, Najmi Varzaneh F, Zare Bidoki A, Rezaei N (2016) Novel WASP mutation in a patient with Wiskott- Aldrich syndrome: Case report and review of the literature. Allergol Immunopathol (Madr 44(5): 450-454.
- Patiroglu T, Klein C, Gungor HE, Ozdemir MA, Witzel M (2016) Clinical Features and Genetic Analysis of Six Patients with Wiskott-Aldrich Syndrome Reporting Two Novel Mutations: Experience of Erciyes University, Kayseri, Turkey. Genet Couns 27(1): 9-24.
- Kamuran K (2017) Wiskott-Aldrich syndrome: Two case reports with a novel mutation. Pediatr Hematol Oncol 34(5): 286-291.
- Shi W, Hou Q, Zhang H, Lou G, Zhang Y (2018) [Analysis of WAS gene mutation in a Chinese family affected with Wiskott-Aldrich syndrome]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 35(2): 207-209.
- Kaneko R, Yamamoto S, Okamoto N, Akiyama K, Matsuno R, et al. (2018) Wiskott-Aldrich syndrome that was initially diagnosed as immune thrombocytopenic purpura secondary to a cytomegalovirus infection. SAGE Open Med Case Rep 6: 2050313X17753788.
- Esmaeilzadeh H, Bordbar MR, Dastsooz H, Silawi M, Fard MF, et al. (2018) A novel splice site mutation in WAS gene in patient with Wiskott-Aldrich syndrome and chronic colitis: a case report. BMC Med Genet 19(1): 123.
- Worth AJ, AJ Thrasher (2015) Current and emerging treatment options for Wiskott-Aldrich syndrome. Expert Rev Clin Immunol 11(9): 1015- 1032.
- Blancas Galicia LC, Escamilla-Quiroz C, Yamazaki-Nakashimada MA (2011) Escamilla-Quiroz, and M.A. Yamazaki-Nakashimada, [Wiskott- Aldrich Syndrome: An updated review]. Rev Alerg Mex 58(4): 213-218.
- Lee PP, & Tong-Xin Chen & Li-Ping Jiang & Jing Chen & Koon-wing Chan, et al. (2009) Clinical and molecular characteristics of 35 Chinese children with Wiskott-Aldrich syndrome. J Clin Immunol 29(4): 490-500.
- Trifari S, Scaramuzza S, Catucci M, Ponzoni M, Mollica L, et al. (2010) Revertant T lymphocytes in a patient with Wiskott-Aldrich syndrome: analysis of function and distribution in lymphoid organs. J Allergy Clin Immunol 125(2): 439-448.
