 Review Article
Review ArticleAbstract
The use of nutraceutical antioxidants in patients with cancer is a controversial debate without any definitive conclusion. Patients affected by cancer take antioxidants during the treatment to alleviate drug toxicities and improve long term outcome. Certain nutraceutical antioxidants could improve the effect of chemotherapy by inducing cells growth arrest, cellular differentiation and by altering redox state of the cells. Many anticancer agents rely on increased oxidative stress for their anticancer activity. On the other hand, the levels of antioxidants could interfere with the effectiveness of chemotherapy by rendering cancer cells less responsive to chemotherapy. Little is known about the use of antioxidants in pediatric cancer. Literature suggests that up to 46% of children with cancer experiences malnutrition during treatment. Antioxidants intake can interfere both with free radical and nutritional status of children with cancer, but it is not clear if it could affect the efficacy of chemotherapy. In Ewing Sarcoma, an aggressive pediatric tumour, the balance between oxidative stress and antioxidants play a central role for the effectiveness of cancer therapy. In this review we propose a literature overview on antioxidants administration during cancer treatment with new original data that identify β3-adrenoreceptor as a potential discriminating factor that could indicate the use of nutraceutical antioxidants in Ewing Sarcoma on the basis of its over expression or down regulation.
Keywords: Biomarkers; Antioxidant; Oxidative stress; Micronutrients; Ewing Sarcoma
Introduction
Nutraceutical antioxidant supplementation in cancer is a controversial and questionable subject, as potential drug-nutrient interactions can affect cancer treatment. Between 13% and 87% of patients with cancer uses antioxidant supplements, often at higher doses compared to those recommended [1,2]. Controversy has been highlighted about the safety of these treatments and the evidences of the efficacy of antioxidant supplement to chemotherapy are indirect and limited [3,4].
The antioxidant nutritional supplements during treatment could alleviate oxidative damage in the healthy tissues, could decrease side effects of treatment and finally could improve the general health and well-being [5-8]. Moreover, tumor response to therapy and patient survival may be improved by the use of antioxidant supplement leading to a better long-term outcome [9,10]. On the other hand, recent literature report that the antioxidant supplementation can affect chemotherapy by reducing its effectiveness. Antioxidants eliminate oxidizing Reactive Oxygen Species (ROS) preventing cellular damage, by affecting antioxidant enzymes by regulating metabolic pathways and providing a adequate supply of non-enzymatic antioxidants [11].
The key enzymatic antioxidant defences are Catalase (CAT), Glutathione Peroxidase (GPx) Superoxide Dismutase (SOD), which constitute the first endogenous defences for the neutralization of ROS. Low amounts of superoxide (O2.-) and hydrogen peroxide (H2O2) are kept by cells, blocking the formation of hydroxyl radicals. Different compounds, eg. Glutathione (GSH), Vitamins A, C and E, Zinc and Selenium represent the non-enzymatic defences [12]. Among the enzymatic antioxidants: SODs convert O2.- to H2O2, catalase reduces H2O2 to water. Moreover, under physiological conditions the reducing power derived from glutathione eliminates H2O2 by molecular oxygen and glutathione peroxidases by the glutathione/glutaredoxin systems. Peroxiredoxin, the Thioredoxin (TRX) are other important mediators of redox signalling in tumor cells [13-19].
When precisely balanced, ROS work as signalling molecules by modulating the activity of the oxidized targets. ROS at low levels promote physiological cells functions by affecting mitogenic proliferation and by acting as second messengers in multiple redox-sensitive signalling reactions [20,21]. The ROS imbalance between pro-oxidant and anti-oxidant processes, either due to excessive ROS production or to decreased scavenging contribute, could lead to an uncontrolled increase of ROS levels that has been considered a common pathway for the onset and progression of many pathologies including cancer, diabetes, neurodegenerative diseases and atherosclerosis. The underlying mechanisms are not yet completely understood. This state of disequilibrium is called ‘oxidative stress’ and can activate all cell pathways involved in cancer progression [12,22]. A “mild” oxidative stress is normally associated with malignant tumour-progression. Cancer cells usually exhibit mitochondrial genomic instability, responsible for metabolic malfunctions and increased production of ROS. Redoxsensitive transcription factors, such as
Nuclear Factor Erythroid 2-Related Factor-2 (NRF2) and the Hypoxia Inducible Factor (HIF-1) once activated, improve antioxidants activities and by increasing cell-survival molecules (Bcl-2 and Akt) [23,24].
The vision of Pediatric Oncologists
Pediatric oncologists have expressed concerns about the supplementation of antioxidants in children with cancer because it could interfere with the anti-tumor activity of conventional therapies [7]. A large proportion of children undergoing treatment for leukaemia, lymphoma and solid tumours suffers from an inadequate growth and a state of malnutrition. The malnutrition in children (6%–50%) depends on the type, stage and location of the tumour [25,26]. For pediatric cancer the debate remains open: should paediatric oncologists provide adequate antioxidant intake, which is essential for normal cellular homeostasis and growth, thereby risking a potential reduction of the effectiveness of anti-cancer therapy, or should not? In this review we evaluate the possible antioxidant supplementations in patients with Ewing Sarcoma (ES). This report will recapitulate available information concerning the pathophysiological role of ROS and antioxidants in cancer and propose a hypothesis concerning a new role of β3-Ars as sensor for the use of antioxidants in Ewing Sarcoma.
Current Data on the use of Nutraceutical Antioxidants
Bioactive nutraceutical compounds with antioxidant ability derived from plants, animals, marine organisms, and microorganisms attracted scientific interest for their beneficial effects on health maintenance. It has been demonstrated that, among natural antioxidant, carotenoids, tocopherols, ascorbates, lipoic acids and polyphenols have strong free radical scavenging activity [27]. Nutraceutical antioxidant supplementation during cancer treatments demonstrated efficacy for many types of malignancies. Interestingly, several anti-tumorigenic drugs derive from natural sources (e.g.: Vincristine, Vinblastine, Paclitaxel, Irinotecan, Topotecan, Etoposide, Daunorubicin, Idarubicin, Actinomycin D) [28,29]. Many bioactive compounds inhibit different pathways involved in tumorigenesis, angiogenesis, and metastasis in pediatric and adult tumors, both in vitro and in vivo by disrupting different cellular enzyme as such as DNA topoisomerase, telomerase. Moreover, antioxidant supplementation interferes with mitotic division, cell-cycle, apoptosis and it eliminates cancer stem cells that are responsible for chemotherapeutic resistance and tumor relapse [30].
Regular consumption of fruits and vegetables reduces the risk of chronic diseases associated with oxidative damage as demonstrated by epidemiological and animal studies [31- 33]. Large-scale epidemiological data reported evidences that treatment with vitamins reduced liver cancer incidence [34]. The supplementation of chlorogenic acid (1µM) suppress glioma growth by affecting M1/M2 macrophage ratio [35]. Interestingly, curcumin (25µM) inhibits the self- glioblastoma stem cell proliferation by inducing intracellular ROS content [36]. In addition, in glioma, combinations of curcumin (0.5µM) and thioridazine (10µM) increase ROS accumulation leading to massive induction apoptosis. In non-small-cell lung cancer Vitamin C treatment exert anti-cancer effects via increasing redox-active labile iron [37] Vitamin E also induce apoptosis in tumour cell lines, increases the efficacy of chemotherapy and reduces drug toxicity [38]. Different vitamin E analogues has been synthesized and screened for their ability to induce human tumour cells to undergo apoptosis [39].
In breast cancer liposome-formulated α-TEA strongly reduced mouse mammary tumour cells growth and lung metastasis in balb/c mice and Vitamin E Succinate (VES) induce DNA synthesis arrest, cellular differentiation and apoptosis leading to cell death [40]. Flavonoids have been reported to have a strong antitumor effect both for antioxidant property and for high cell death induction.
It has been reported that flavonoid apigenin reduces hepatocellular carcinogenesis via decreasing oxidative stress and it exerts anti-tumor effects in various types of cancers, including colorectal cancer, breast cancer, liver cancer, lung cancer, melanoma, prostate cancer and osteosarcoma [41-47]. Apigenin disrupts tumor angiogienesis by revealing similar activity of Axitinib (angiogenesis inhibitor) exerting the same binding activity to the VEGF receptors including VEGFR1 and VEGF R2 [48]. Moreover, this flavone directly inhibits cancer cell proliferation by inducing apoptosis, autophagy and cell cycle arrest. It can reduce oxidative stress, increase hepatic detoxification enzyme efficacy, and act as anti-inflammatory [49,50]. Several studies also reported the synergistic effect of genistein when administered together with other anticancer
drugs by inhibiting MAPK activation and mediating trail induced apoptosis [51]. Despite the great interest of these preclinical data, there are no consistent data regarding the use of flavonoids in human as anticancer therapy. Epidemiologic and clinical trials revealed inconsistent results about the clear dietary recommendations for the intake of flavonoids to support human health [52].
It has been reported that retinoic acid suppress prostate, breast, lung, ovarian, bladder, oral, and skin cancers by causing cell cycle arrest through the inhibition of p27 and Cdk5 and by increasing apoptosis of cancer cells [53]. In pancreatic cancer therapy, combination therapy of gemcitabine with trichostatin A, Epigallocate-3-Gallate (EGCG), capsaicin and Benzyl Isothiocyanate (BITC) have been proven as effective. In addition, Aminoflavone (5-amino-2-(4-amino-3-fluorophenyl)- 6,8-difluoro-7-methylchromen-4-one; AF) is highly selective as inductor of cell death as reported by its ability to kill MCF-7 and MDA-MD-468 breast cancer cells but not non-malignant MCF10A breast epithelial cells. Aminoflavone activates caspase 3 and increases intracellular ROS levels correlating with induction of apoptosis [54,55]. Literature reported that prooxidant toxicity of flavonoids is considered the principal mechanisms by which they inhibit mitochondrial breathing. Cu2+ and Fe2+ which are present in biological systems modulate prooxidant activity of flavonoids, by improving selective cytotoxicity in cancer cells that contain more copper compared with normal cells [56,57].
However, opposites studies keep increasing evidence that the scavenging effect of ROS is in fact deleterious to cancer patients rather than preventing the risk. In another study, N-Acetylcysteine (NAC), a medication that works by increasing the glutathione levels and vitamin E, was shown to accelerate the tumor progression in a B-RAF and K-RAS induced lung cancer in murine models. These antioxidants promote tumour progression by reducing ROS levels and in turn reduce p53 expression levels that could have induced apoptosis leading to the cell death [58]. The role of antioxidants on fighting ROS damage in tumor cells has been implicated in chemoresistance and poor overall survival of cancer patients. Elevated levels of GSH has been indicated as unfavorable factor for the sensitivity to chemotherapeutic agent, as well as high levels of SOD-2 contribute to cancer progression [59,60]. Vitamin A (25,000 IU) supplement leads to an increased risk of lung cancer changing redox homeostasis states in different cancer cell types [61]. In addition, several other investigations have demonstrated that anti-oxidant nutraceuticals accelerate cancer progession by rendering the tumor cells insensitive to elevation of ROS induced by chemotherapic agent [62,63].
Understanding the Controversy
The published literature regarding concurrent chemotherapy or radiation with antioxidants reaches a wide variety of conclusions, some showing improved survival and status and others a reduced survival. Many questions remain open: “Do antioxidants affect the efficacy of anticancer agents by improving toxicity in cancer cells? Do antioxidants protect cancer cells from the effect of chemotherapy?” [31]. The controv ersial role of antioxidants during cancer therapy could depend on the antioxidants dose used: a prophylactic or a therapeutic dose, ie. a low or elevated dose respectively. Prophylactic dose protects healthy cells and tumour cells. On the contrary, a therapeutic dose inhibits the growth of cancer cells but not of healthy cells. On the other hand, higher levels of endogenous antioxidants could adverse oxidative stress induced by chemotherapeutic agents especially in younger patients having impaired capacity to deal with oxidative insult due an increased level of malnutrition status [3,64]. Definitely, dietary antioxidant supplements can be considered as “double-edged sword” in cancer treatment, for their ability either to kill cancer cells or protect them.
The Redox State in Ewing’s Sarcoma Cells
Ewing`s Sarcoma Family Tumors (ESFTs) are round cell tumors that develop in bone and soft tissues of children and young adults [65]. It is a quite common paediatric malignant tumour, with a frequency of 2% of all childhood cancers [66]. Until now combined chemotherapy regimen, radiotherapy and surgery, remains the only strategy to overcome the disease. At diagnosis, approximately 25- 30% of patients with ES have metastatic disease [67]. It presents high incidence of local or distant relapse, up to 40% in metastatic setting. Unfortunately, approximately 20-30% of patients with metastatic or recurrent disease have a poor prognosis even undergoing intensive multi-drug chemoterapy regimen [68]. For patients with localized disease, an intensive multimodal treatment (chemotherapy combined with surgery and/or radiotherapy), achieves a survival rate of approximately 70%. The 10-year survival rate for patients with metastatic disease increased from 16 to 30% after the introduction of multidrug chemotherapy.
Standard chemotherapy includes Adriamicin, Vincristin, Cyclophosphamide and Etoposide. For recurrent disease, only few chemotherapy agents as topotecan and cyclophosphamide, ifosfamide in combination with carboplatin and etoposide, irinotecan and temozolamide showed activity with modest but shortlasting response rate: Patients with metastatic disease often present resistance to chemotherapeutic agents [69].
The origin of ES remain controversial since no precancerous lesions have been described in literature. It is now reported to origin from mesenchymal and neural crest. Over 90% of ES cases present the EWS-FLI1 fusion protein derived from recurrent reciprocal translocations that connect the EWS gene on chromosome 22 with an ETS family gene, either FLI-1 on chromosome 11 or ERG on chromosome 21 [70,71]. EWS-FLI fusion protein is an aberrant protein working as sequence-specific transcription factor leading to neoplastic transformation and tumor progression. Moreover, ES expresses CD99 surface marker, also present in Synovial Sarcoma (SS) and Low-Grade Fibromyxoid Sarcoma (LGFMS). The down regulation of CD99 in human ES cell lines reduced their ability to form tumors and bone metastases in immunodeficient mice and, in vitro, decreased their tumorigenic and metastatic features [72].
Novel therapeutic approaches are needed for metastatic patients resistant to chemotherapy [73]. Preclinical and clinical studies reported drugs targeting EWS-FLI1 fusion protein as small-molecule YK-4-279 that inhibit interaction of EWS-FLI1 and RNA helicase A. Moreover, it has been developed new therapeutic strategy targeting CD99, Vascular Endothelial Growth Factor (VEGF) and its receptor, insulin-like Growth Factor-1 (IGF1) pathways and Mammalian Target of Rapamycin (mTOR), osteoclastic-osteoblastic homeostasis and bone microenvironment, enzymatic pathways (poly ADP- ribose polymerase 1 - PARP1), and GD2 ganglioside pathways [74-78].
Metabolic activity of ES is abnormal with an increased glucose uptake and activation of accelerated glycolysis that provides energy demanded from cancer cells, thus leading to a dysfunction in oxidative phosphorylation activities and redox state imbalance. Therefore, the intracellular redox environment changes rapidly in ES cells. The basis for the growth or the reduction of ES tumour is represented by regulation of ROS production and detoxification balance. Pharmacological approach that decrease cellular antioxidant activity, increasing oxidative stress and inducing of cell death represents one of the most important therapeutic strategy in ES. The antioxidant inhibition strategy has been proposed to enhance the efficacy of chemotherapy as doxorubicin and etoposide which increase generation of ROS and oxidative stress in mitochondria [79]. The majority of conventional drugs used in ES affects ROS balance by increasing the level of ROS over the toxic threshold to kill cancer cells. ES cell lines and patients present different response to the induction of cell death by chemotherapic agents depending on different levels of intracellular antioxidants and the different ability to neutralise ROS [80].
Cellular antioxidants are differentially expressed in ES cell lines. GSH in ES plays an important antioxidant role, therefore, depletion of intracellular GSH enhances the efficacy of ROS-production induced by anticancer agents such as fenretinide leading to a massive cell death [80]. In ES the GSH metabolism pathway altered by upregulation of g-glutamyltranspeptidase enzyme is associated with poor prognosis [81,82]. GSH influences multiple cellular processes, including proliferation, cell differentiation and apoptosis, and its altered homeostasis is involved in the aetiology and progression of different type of cancers and other pathologies. In many tumor cells, elevated GSH levels increase the antioxidant ability and the resistance to oxidative stress while the decrease in the GSH/ GSSG ratio, leads to an increased susceptibility to oxidative stress leading to cancer onset and progression. ES cell lines have high GSH/GSSG ratio that maintain a high basal oxidative stress level which increases the expression of different antioxidants [80].
Recent studies in ES have shown that the Glutathione S-Transferases (GSTs), a family of detoxification enzymes, are direct targets for EWS-FLI1 [83]. GSTs are involved in the development of resistance to cancer chemotherapeutic agents by detoxifying many compounds such as doxorubicin and vincristine [59]. Tumors show high levels of GST expression compared to normal tissues. Over expression of MGST4 (membrane-bound GST4) and MGST1 (membrane-bound GST1) predicts a poor response to chemotherapy as doxorubicin and are associated with poor prognosis [81,83]. MGST1, expressed in the endoplasmic reticulum and outer mitochondrial membrane, protects cells and mitochondria maintaining the homeostasis of glutathione peroxidase function [84]. In Ewing sarcoma cells (SKN-MC) catalase inhibition could represent a potential strategy to increase the sensitivity of ES to chemotherapeutic agents [85]. Various strategies have been studied to increase ROS content in ES cells but none of these proved to be efficacious due to an increased antioxidant response overcoming the activity of chemotherapy. Usually, chemotherapeutic agents increase antioxidants as response to mild levels of ROS, rendering the cells less sensitive to chemotherapy.
New Insight: β3-AR as Prosurvival Factor under Nutraceutical Antioxidant Treatment
Recently, β3 adrenergic receptor (β3-AR) becomes incredibly attractive in cancer biology. Literature reports data about the ability of β3-AR to reduce tumour growth and metastases. Hypoxic induction of β3-AR has been recently reported in tumor microenvironment and overexpression of β3-AR has been associated with cancer growth, recruitment of circulating stromal cell precursors to the tumor sites and enhancement of stem cell traits [86]. The β3-AR antagonist, SR59230A, reduces cell proliferation and induces apoptosis in mouse B16F10 melanoma cells, through a mechanism mediated by inducible isoform of nitric oxide synthase and reduces angiogenesis by inhibiting VEGF secretion. In tumor bearing mice melanoma cell proliferation and tumor vasculature are reduced by intra-tumor injections of SR59230A thus resulting in significant decrease in tumor growth [87].
β3-AR is expressed in bladder, in lymphocytes (at a low level) and in White and Brown Adipose Tissue (WAT, BAT), where its role has been well clarified. β3-AR are involved in adipose tissue morphology and metabolism, administration of selective β3- AR agonist CL- 316,243 increases thermogenesis mediated by Uncoupling Protein 1 (UCP-1) in BAT and lipolysis in WAT [88]. UCPs maintain redox state of the cells in the respiratory chain transport. Over the past decade β3-AR has been proposed as a potential therapeutic target in several pathologies such as cachexia, obesity, diabetes, cardiac disease and in over-active bladder [89], and recent literature reported a new role of β3-AR on cancer progression and dissemination [87].
Interestingly, UCP2 has been shown to control GSH/GSSG in beta pancreatic cells [90]. In particular, as we previously showed in melanoma, β3-AR is expressed on mitochondria driving the activity of UCP-2 and piloting ROS content in the mitochondria [91]. More recently, it has been reported a dual role of β3-AR on antioxidant cell response, it directly inhibits NADPHox activity and induces the expression of catalase. Literature also reported that noradrenaline induces catalytic subunit of Glutamate-Cysteine Ligase Protein (GCLc) thus increasing the intracellular GSH level through stimulation of β3-AR in U-251 MG cells and in mouse astrocytes in primary culture. In this work is reported that β3-AR induces GSH synthesis in glioma cells thus maintaining GSH homeostasis [92,93]. Interestingly, most of the nutraceutical compounds increase β3-AR levels, potentially driving an antioxidant response and cell survival trough activation of ERK or inhibiting the apoptotic process.
In 1997 was revealed that there is connections between ROS production and UCPs activity in experiments where an inhibitor of UCP1, GDP, increases ∆ψ and ROS production [94]. It has also been reported that ROS production and their levels are correlated with UCPs activation superoxide mediated [95]. Administration of several nutraceutical compounds differentially affect cell survival and apoptosis in ES cells. Specifically, here we report that the ES tumor cells A673 treated with Curcumin, Retinoic Acid, 8-Gingerol and Genistein exhibited reduced viability if compared with A673 cells treated with Capsaicin, Ascorbic Acid, Formononetin and Flavon where the treatment did not affect cell viability (Figures 1A&1B). Moreover, the intracellular expression of mitochondrial ROS remained at low levels in ES cells treated with prosurvival antioxidants and increased in the cells treated with antioxidants that are able to reduce cell viability (Figures 2A&2B). Interestingly, the treatments that that do not affect cell viability up regulated the expression of β3-AR and the treatment that decreased cell viability strongly down regulated β3-AR (Figures 3A&3B).
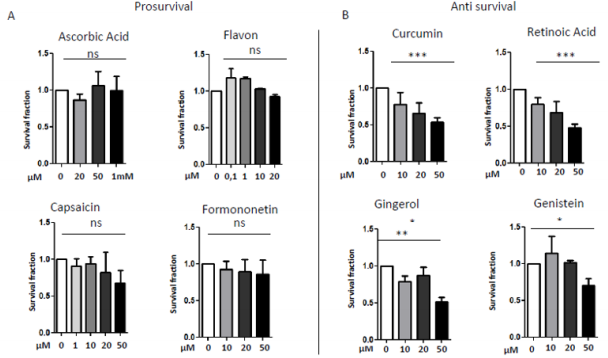
Figure 1: MTT survival analysis in A673 ES cells treated with different nutraceutical; A) Treatments with Ascorbic Acid (20µM, 5 µM, 1mM), Flavon (100nM, 1µM, 10µM, 20µM), Capsaicin (1µM, 10µM, 20µM, 50µM), Formononetin (10µM, 20µM, 50µM) that did not affect cells viability. B) Treatments with Curcumin (10µM, 20µM, 50µM), Retinoic Acid (10µM, 20µM, 50µM), Gingerol (10µM, 20µM, 50µM) and Genistein (10µM, 20µM, 50µM), that reduced cells viability. NS: not significant. P values for treatments: ∗P <0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
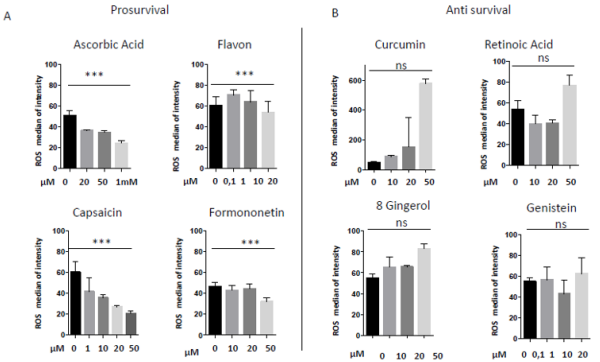
Figure 2: Mitochondrial ROS measurement in A673 ES cells treated with different nutraceuticals; A) Decrease of mtROS levels after treatments with Ascorbic Acid (20µM, 50µM, 1mM), Flavon (100nM, 1µM, 10µM, 20µM), Capsaicin (1µM, 10µM, 20µM, 50µM), Formononetin (10µM, 20µM, 50µM). B) Increase of mtROS levels after treatments with Curcumin (10µM, 20µM, 50µM), Retinoic Acid (10µM, 20µM, 50µM), Gingerol (10µM, 20µM, 50µM) and Genistein (10µM, 20µM, 50µM). NS: not significant. P values for treatments: ∗∗∗P < 0.001.
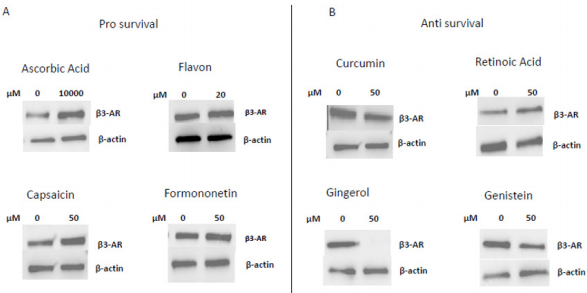
Figure 3: Western Blotting analysis of β3-AR expression after treatment with different nutraceuticals. A) Up-regulation of β3-AR expression after treatment with nutraceuticals that do not affect cells viability: Ascorbic Acid 1mM, Flavon 20µM, Capsaicin 50µM, Formononetin 50µM. B) Down regulation of β3-AR levels after treatment with nutraceuticals that reduce cells viability: Curcumin 50µM, Retinoic Acid 50µM, Gingerol 50µM and Genistein 50µM. β actin is shown as loading control.
Since β3-AR is expressed in the mitochondria, it could drive mitochondrial bioenergetics and the redox state of the cells driving or not cell death under nutraceuticals treatment. Here we identified the β3-AR receptor as the main regulator of the cellular response to oxidative stress in the cells under different micronutrients treatment. It works as sensor for ROS control in ES cells by driving or not cells antioxidant response and cell death. Since β3- AR antagonism lead to massive cell death, inhibiting β3-AR in these cells could dramatically increase the ROS levels by toxic threshold leading to cell death by inhibiting the antioxidant response of the cells.
Since we previously reported that β3-AR antioxidant activity is mediated by UCP-2 protein expression in melanoma, it works as mediator for the elevation of endogenous antioxidants activity of the cells. Here, we report that β3-AR could control the redox state of the cells working as ROS sensor, driving cells to life or to death. It could be possible that this activity is linked to mitochondria bioenergetics function.
Materials and Methods
Cell Cultures
Human Ewing Sarcoma (ES) cells A673 were cultured in 100mm plates in DMEM high glucose medium (4.5 g/L) supplemented with 10% Fetal Bovine Serum (FBS), 5% of L- glutammin, 5% of penicillin and streptomycin (1%) and were mantained at 37°C in a 5% CO2 humidified atmosphere incubator. Cells were usually stored in liquid nitrogen in a freezing solution, containing 95% complete DMEM and 5% Dimethyl Sulfoxide (DMSO) and then plated in petri p100. For defrosting, the vials were rapidly brought to 37°C by immersion in the thermostat bath, then centrifuged to remove the toxic DMSO from the cells, re-suspended in DMEM high glucose FBS 10% and appropriately plated. Sub-confluent cells were detached from plate with trypsin-enzyme after aspirating the medium and after one wash with the Phosphate Buffered Saline (PBS) to eliminate medium and serum residues. Then DMEM high glucose was added and the cell suspension obtained was counted and plated in fresh DMEM high glucose with appropriate dilutions. Hypoxic experiment were performed in hypoxic incubator at 1% of O2 atmosphere.
Cell Treatments
A673 ES cells were plated to reach 70% confluence in complete high glucose DMEM medium. After 24h the medium was removed, the cells were washed in PBS solution, and finally starved overnight with starvation medium, without FBS, in order to promote cell’s entry into a quiescent G0 phase, thereby better evaluating cells responsiveness to exogenous treatments. The consequent morning, cells were treated with a single dose of Apigenin at the concentrations of 10, 20, 50 µM and subsequently left in incubator for 24h, then collected for the experiments. Apigenin was dissolved in dymethylsulphoxide 2,7 mg/ml to obtain a final concentration stock of 10mM, then appropriate dilutions from the stock solution were made for treatments.
MTT Assay
The effect of Apigenin on cell viability was detected by MTT assay. A673 cells were transferred into a 96-wells plate at a density of 10 x 10³ cells/well in 150µl DMEM complete. A673 cells were incubated with various doses of Apigenin (10µM, 20µM, 50µM) for 24 and 48h separetely. A total of 10µl of MTT was added to each well and incubated under darkness for 1h at 37°C. Then culture medium was removed and 150µl of DMSO was added to each well. The intensity of absorbance was detected at 570nm using a dualbeam microplate reader.
Glutathione Fluorometric Assay Kit
For the detection of reduced Glutathione (GSH) the Glutathione Fluorometric Assay Kit (BioVision) was used, following the manufacturer instructions.
MitoSOX and Live/Dead Cells Assays
For the intracellular ROS measurements, A673 cells plated into 24-wells plates at a density of 10 x10⁴ cells in 1ml complete high glucose DMEM medium, were treated with Apigenin as described above. After 3, 6, 24h of treatment, cells were stained with 1µl of MitoSOX reagent at concentration of 2.5µM. After 15 minutes of incubation at room temperature under darkness, cells were washed with PBS, detached with 250µl of accutase buffer, spinned at 1300 rpm for 5 minutes and pellet was resospended in 101µl of live/ dead cells mix (Viobility 488/520 Fixable Dye previously prepared following the protocol). Cells were incubated under darkness for 10 minutes and were washed with 1ml of PBS, spinned at 1300 rpm for 5 minutes and pellet was resospended in 300µl of PBS and then evaluated by flow citometry for MitoSOX-PE reagent.
Western Blot
After homogenization and protein quantifiation, samples were subjeceted to SDS-PAGE and Western Blot analysis. Subsequently PVDF membranes were incubated for 1 hour in slow agitation at room temperature in a blocking solution of non-fat dry milk 2% and Tween PBS 0,05% in order to avoid the formation of unspecific ties. Membranes were then incubated with primary antibody: β3-adrenergic receptor (Abcam), Catalase (Abcam), Superoxide dismutase-2 (Santa Cruz), Txnip (Life Technologies), β-actin (Santa Cruz). The primary antibody was added generally in a concentration of 1:1000 and incubated, in shaking, over- night at 4°C. The next day membranes were washed th ree times with a washing solution containing PBS 1X and Tween 0.1% in order to remove unbound primary antibody in excess. Then, the specific secondary antibody, which was conjugated with Horseradish Peroxidase (HPR), was added, in a dilution of 1:5000 in milk and incubated for 1 hour. Chemiluminescent protein’s revelation was carried out with ECL reagent and developing of blots was carried out by Chemidoc Imaging System (Bio-Rad). To verify the application of equal amounts of protein, the intensity of the corresponding protein bands of interest was normalized on the β-actin bandfor each sample.
Statistical Analysis
in vitro data are presented as means ± Standard Deviation (SD) from at least three experiments. Results were normalized versus control expression levels. Statistical analysis was performed using Graph Pad Prism software (GraphPad, San Diego CA, USA) by OneWay Analysis Variance (ANOVA), followed by Bonferroni post hoc analysis.
Conclusion
in vitro and in vivo data suggest that many nutraceutical antioxidants can enhance or reduce the effects of cytotoxic therapy by differentially altering the intracellular redox state and can promote cells proliferation and malignant progression. Therefore, the use of antioxidants especially in paediatric patients, which are constantly under uncertain nutritional status, remains questionable.
In this review we hypothesize that supplement of determined antioxidants and nutrients can contribute to raise the health status of young patients affected by ES only when nutraceutical supplementation inhibits β3-AR expression. Could antioxidants and β3-AR antagonists be helpful in Ewing Sarcoma? SR5920A could increase oxidative stress clarifying the way for the use of antioxidants to kill cancer cells and maintain the wellness of normal cells where this receptor is expressed at a lower level. Moreover, we showed that SR59230A strongly reduces cancer cell viability, demonstrating that blocking of β3-AR function could represent a novel therapeutic strategy for ES through its ability to reduce antioxidant activity [96].
Acknowledgment
Thanks to Margherita Nardi and Marina Vignoli for kindly revised the manuscript.
References
- Vande Creek L, Rogers E, Lester J (1999) Use of alternative therapies among breast cancer outpatients compared with the general population. Altern Ther Health Med 5(1): 71-76.
- Block KI, Koch AC, Mead MN, Tothy PK, Newman RA, et al. (2008) Impact of antioxidant supplementation on chemotherapeutic toxicity: A systematic review of the evidence from randomized controlled trials. Int J Cancer 123(6): 1227-1239.
- Singh K, Bhori M, Kasu YA, Bhat G, Marar T (2018) Antioxidants as precision weapons in war against cancer chemotherapy induced toxicityExploring the armoury of obscurity. Saudi J Pharmac 26: 177-190.
- Velicer CM, Ulrich CM (2008) Vitamin and mineral supplement use among US adults after cancer diagnosis: A systematic review. J Clin Oncol 26: 665-673.
- Norman H, Butrum R, Feldman E, Heber D, Nixon D, et al. (2003) The role of dietary supplements during cancer therapy. J Nutr 133: 3794S-3799S.
- Greenlee H, Kwan M, Kushi L, Song J, Castillo A, et al. (2012) Antioxidant supplement use after breast cancer diagnosis and mortality in the Life After Cancer Epidemiology (LACE) cohort. Cancer 118: 2048-2058.
- Ladas EJ, Jacobson JS, Kennedy DD, Teel K, Fleischauer A, et al. (2004) Antioxidants and cancer therapy: A systematic review. J Clin Oncol 22: 517-528.
- Lawenda B, Kelly K, Ladas E, Sagar S, Vickers A, et al. (2008) Should supplemental antioxidant administration be avoided during chemotherapy and radiation therapy? J Natl Cancer Inst 100: 773-783.
- Conklin K (2000) Dietary antioxidants during cancer chemotherapy: Impact on chemotherapeutic effectiveness and development of side effects. Nutr Cancer 37: 1-18.
- Ladas E, Kelly KM (2010) The antioxidant debate. Explore (NY) 6(2): 75-85.
- He L, He T, Farrar S, Ji L, Liu T, et al. (2017) Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol Biochem 44(2): 532-553.
- Liou GY, Storz P (2010) Reactive oxygen species in cancer. Free Radic Res 44(5): 479-496.
- Lu J, Holmgren A (2014) The thioredoxin antioxidant system. Free Radic Biol Med 66: 75-87.
- Hanschmann EM, Godoy JR, Berndt C, Hudemann C, Liling CH (2013) Thioredoxins, glutaredoxins, and peroxiredoxins-molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antiox Redox Signal 19: 1539-1605.
- Du Y, Zhang X, Lu J, Holmgren A (2013) Thioredoxin 1 is inactivated due to oxidation induced by peroxiredoxin under oxidative stress and reactivated by the glutaredoxin system. J Biol Chem 288: 32241-32247.
- Brautigam L, Jensen LD, Poschmann G, Nystrom S, Bannenberg S, et al. (2013) Glutaredoxin regulates vascular development by reversible glutathionylation of sirtuin 1. Proc Natl Acad Sci USA 110: 20057-20062.
- Peskin AV, Pace PE, Behring JB, Paton LN, Soethoudt M, et al. (2016) Glutathionylation of the active site cysteines of peroxiredoxin 2 and recycling by glutaredoxin. J Biol Chem 291: 3053-3062.
- Chen JW, Dodia C, Feinstein SI, Jain MK, Fisher AB (2000) 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. J Biol Chem 275: 28421-28427.
- Day AM, Brown JD, Taylor SR, Rand JD, Morgan BA, et al. (2012) Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Mol Cell 45: 398-408.
- Helfinger V, Schröder K (2018) Redox control in cancer development and progression. Mol Aspects Med 63: 88-98.
- Di Meo S, Reed TT, Venditti P, Victor VM (2016) Role of ROS and RNS sources in physiological and pathological conditions. Oxid Med Cell Longev 2016: 1245049.
- Pham Huy LA, He H, Pham Huy C (2008) Free radicals, antioxidants in disease and health. In J Biomed Sci 4(2): 89-96.
- Dengler LV, Galbraith M, Espinosa JM (2014) Transcriptional regulation by hipoxia inducible factors. Crit Rev Biochem Mol Biol 49(1): 1-15.
- Calvani M, Comito G, Giannoni E, Chiarugi P (2012) Time-dependent stabilization of hypoxia inducible factor-1α by different intracellular sources of reactive oxygen species. PLoSOne 7(10): e38388.
- Donaldson SS, Wesley MN, DeWys WD, Suskind RM, Jaffe N (1981) A study of the nutritional status of pediatric cancer patients. Am J Dis Child 135(12): 1107-1112.
- Van Eys J (1979) Malnutrition in children with cancer: Incidence and consequence. Cancer 43(5 Suppl): 2030-2035.
- Watkins R, Wu L, Zhang C, Davis RM, Xu B (2015) Natural product-based nanomedicine: Recent advances and issues. Int J Nanomedicine 10: 6055-6074.
- Rocha AB, Lopes RM, Schwartsmann G (2001) Natural products in anticancer therapy. Curr Opin Pharmacol 1(4): 364-369.
- Mann J (2002) Natural products in cancer chemotherapy: Past, present and future. Nat Rev Cancer 2(2): 143-148.
- Ferrucci V, Boffa I, De Masi G, Zollo M (2006) Natural compounds for pediatric cancer treatment. Naunyn Schmiedebergs Arch Pharmacol 389(2): 131-149.
- Rohenkohl CC, Carniel AP, Colpo E (2011) Antioxidants consumption during chemotherapy treatment. ABCD Arq Bras Cir Dig 24(2): 107-112.
- Boeing H, Bechthold A, Bub A, Ellinger S, Haller D, et al (2012) Critical review: Vegetables and fruit in the prevention of chronic diseases. Eur J Nutr 51(6): 637-663.
- Key TJ (2011) Fruit and vegetables and cancer risk. Br J Cancer 104(1): 6-11.
- Zhang W, Shu XO, Li H, Yang G, Cai H, et al (2012) Vitamin intake and liver cancer risk: A report from two cohort studies in China. J Natl Cancer Inst 104(15): 1173-1181.
- Xue N, Zhou Q, Ji M, Jin J, Lai F, et al (2017) Chlorogenic acid inhibits glioblastoma growth through repolarizating macrophage from M2 to M1 phenotype. Sci Rep 7: 39011.
- Gersey ZC, Rodriguez GA, Barbarite E, Sanchez A, Walters WM, et al. (2017) Curcumin decreases malignant characteristics of glioblastoma stem cells via induction of reactive oxygen species. BMC Cancer 17: 99.
- Schoenfeld JD, Sibenaller ZA, Mapuskar KA, Bradley MD, Wagner BA, et al. (2018) Redox active metals and H2O2 mediate the increased efficacy of pharmacological ascorbate in combination with gemcitabine or radiation in pre-clinical sarcoma models. Redox Biol 14: 417-422.
- Lamson DW, Brignall MS (1999) Antioxidants in cancer therapy; their actions and interactions with oncologic therapies. Altern Med Rev 4(5): 304-29.
- Kline K, Yu W, Sanders BG (2004) Vitamin E and breast cancer. J Nutr 134(12 Suppl): 3458S-3462S.
- Schwenke DC (2002) Does lack of tocopherols and tocotrienols put women at increased risk of breast cancer? J Nutr Biochem 13(1): 2-20.
- Jeyabal PV, Syed MB, Venkataraman M, Sambandham JK, Sakthisekaran D (2005) Apigenin inhibits oxidative stress-induced macromolecular damage in N-nitrosodiethylamine (NDEA)-induced hepatocellular carcinogenesis in Wistar albino rats. Mol Carcinog 44(1): 11-20.
- Xu M, Wang S, Song YU, Yao J, Huang K, et al. (2016) Apigenin suppresses colorectal cancer cell proliferation, migration and Invasion via inhibition of Wnt/beta-catenin signaling pathway. Oncol Lett 11: 3075-3080.
- Huang C, Wei YX, Shen MC, Tu YH, Wang CC (2016) Chrysin, abundant in Morindacitrifolia fruit water-EtOAc extracts, combined with apigenin synergistically induced apoptosis and inhibited migration in human breast and liver cancer cells. J Agric Food Chem 64: 4235-4245.
- Lee YM, Lee G, Oh TI, Kim BM, Shim DW, et al. (2016) Inhibition of glutamine utilization sensitizes lung cancer cells to apigenin-induced apoptosis resulting from metabolic and oxidative stress. Int J Oncol 48: 399-408.
- Zhao G, Han X, Cheng W, Ni J, Zhang Y, et al. (2017) Apigenin inhibits proliferation and invasion and induces apoptosis and cycle arrest in human melanoma cells. Oncol Rep 37: 2277-2285.
- Gupta S, Afaq F, Mukhtar H (2002) Involvement of nuclear factor-kappa B, Bax and Bcl-2 in induction of cell cycle arrest and apoptosis by apigenin in human prostate carcinoma cells. Oncogene 21: 3727-3738.
- Angulo P, Kaushik G, Subramaniam D, Dandawate P, Neville K, et al (2017) Natural compounds targeting major cell signaling pathways: a novel paradigm for osteosarcoma therapy. J Hematol Oncol 10: 10.
- Seo EJ, Kuete V, Kadioglu O, Krusche B, Schröder S, et al. (2013) Antiangiogenic activity and pharmacogenomics of medicinal plants from traditional korean medicine. Evid Based Complement Alternat Med 2013: 131306.
- O’Prey J, Brown J, Fleming J, Harrison PR (2003) Effects of dietary flavonoids on major signal transduction pathways in human epithelial cells. Biochem Pharmacol 66(11): 2075-2088.
- Surh YJ (2003) Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer 3(10): 768-80.
- Spagnuolo C, Russo GL, Orhan IE, Habtemariam S, Daglia M, et al. (2015) Genistein and cancer: current status, challenges, and future directions. Adv Nutr 6(4): 408-419.
- Balentine DA, Dwyer JT, Erdman JW, Ferruzzi MG, Gaine PC, et al. (2015) Recommendations on reporting requirements for flavonoids in research. Am J Clin Nutr 101(6): 1113-25.
- Chen MC, Hsu SL, Lin Ho, Yang TY (2014) Retinoic acid and cancer treatment. Biomedicine 4(4): 22.
- Zhang R, Humphreys I, Sahu RP, Shi Y, Srivastava SK (2008) in vitro and in vivo induction of apoptosis by capsaicin in pancreatic cancer cells is mediated through ROS generation and mitochondrial death pathway. Apoptosis 13(12): 1465-1478.
- Callero MA, Loaiza-Pèrez Al (2011) The role of aryl hydrocarbon receptor and crosstalk with estrogen receptor in response of breast cancer cells to the novel antitumor agent benzothiazoles and aminoflavone. In J Breast Cancer 2011: 923250.
- Eghbaliferiz S, Iranshahi M (2016) Prooxidant activity of polyphenols, flavonoids, anthocyanins and carotenoids: Updated review of mechanisms and catalyzing metals. Phytother Res 30: 1379-1391.
- Perron NR, Brumaghim JL (2009) A review of the antioxidant mechanisms of polyphenol compounds related to iron binding. Cell Biochem Biophys 53: 75-100.
- Sarangarajan R, Meera S, Rukkumani R, Sankar P, Anuradha G (2017) Antioxidants: Friend or foe? Asian Pac J Trop Med 10(12): 1111-1116.
- Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, et al. (2013) Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev 2013: 972913.
- Yi L, Shen H, Zhao M, Shao P, Liu C, et al. (2017) Inflammationmediated SOD-2 upregulation contributes to epithelial -mesenchymal transition and migration of tumor cells in aflatoxin G1-induced lung adenocarcinoma. Sci Rep 7(1): 7953.
- Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, et al. (1996) Risk factors for lung cancer and for intervention effects in CARET, the beta-carotene and retinol efficacy trial. J Natl Cancer Inst 88(21): 1550-1559.
- Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, et al. (2014) Antioxidants accelerate lung cancer progression in mice. Sci Transl Med 6(221): 221.
- Stebbing J, Hart CA (2011) Antioxidants and cancer. Lancet Oncol 12(11): 996.
- Conklin KA (2004) Cancer chemotherapy and antioxidants. J Nutr 134(11): 3201S-3204S.
- Kovar H (1998) Ewing’s Sarcoma and peripheral primitive neuroectodermal tumors after their genetic union. Curr Opin Oncol 10(4): 334-342.
- Riggi N, Stamenkovic I (2007) The Biology of Ewing sarcoma. Cancer Lett 254(1): 1-10.
- Balamuth NJ, Womer RB (2010) Ewing’s sarcoma. Lancet Oncol 11(2): 184-192.
- Grünewald TG P, Cidre-Aranaz F, Surdez D, Tomazou EM , de Alva E, et al. (2018) Ewing sarcoma. Nat Rev Dis Primers 4(1): 5.
- Jain S, Kapoor G (2010) Chemotherapy in Ewing’s sarcoma. Indian J Orthop 44(4): 369-377.
- Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, et al. (1992) Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumors. Nature 359(6391): 162-5.
- Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, et al. (1994) A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS- family transcription factor, ERG. Nat Genet 6(2): 146-151.
- Pasello M, Manara MC, Scotland K (2018) CD99 at the crossroads of physiology and pathology. J Cell Commun Signal 12(1): 55-68.
- Ozaki T (2015) Diagnosis and treatment of Ewing of the bone: A review article. J Orthop Sci 20(2): 250-263.
- DuBois SG, Marina N, Glade-Bender J (2010) Angiogenesis and vascular targeting in Ewing sarcoma: A review of preclinical and clinical data. Cancer 116(3): 749-757.
- Lamhamedi-Cherradi SE, Menegaz BA, Ramamoorthy V, Vishwamitra D, Wang Y, et al. (2016) IGF-1R and mTOR blockade: Novel resistance mechanisms and synergistic drug combinations for ewing sarcoma. J Nat Cancer Inst 108(12): 182.
- Kailayangiri S, Altvater B, Meltzer J, Pscherer S, Leucke A, et al. (2012) The ganglioside antigen G(D2) is surface-expressed in Ewing sarcoma and allows for MHC- independent immune targeting. Br J Cancer 106(6): 1123-1133.
- Mancarella C, Scotlandi K (2018) IGF system in sarcomas: A crucial pathway with many unknowns to exploit for therapy. J Mol Endocrinol 61(1): T45-T60.
- Vormoor B, Curtin NJ (2014) Poly(ADP-ribose) polymerase inhibitors in Ewing sarcoma. Curr Opin Oncol 26(4): 428-433.
- Zhou S, Starkov A, Froberg MK, Leino RL, Wallace KB (2001) Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res 61(2): 771-777.
- Magwere T, Myatt SS, Burchill SA (2008) Manipulation of oxidative stress to induce cell death in Ewing’s sarcoma family of tumors. Eur J Cancer 44(15): 2276-2287.
- Scotlandi K, Remondini D, Castellani G. et al (2009) Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J Clin Oncol 27(13): 2209-2216.
- Bouman L, Sanceau J, Rouillard D, Bauvois B (2002) γ-glutamyltranspeptidase expression in Ewing’s sarcoma cells: upregulation by interferons. Biochem J 364: 719-724.
- Luo W, Gangwal K, Sankar S (2009) GSTM4 is a microsatellite-containing EWS/FLI target involved in Ewing’s sarcoma oncogenesis and therapeutic resistance. Oncogene 28(46): 4126-4132.
- Johansson K, Jarvliden J, Gogvadze V, Morgenstern R (2010) Multiple roles of microsomal glutathione transferase 1 in cellular protection: a mechanistic study. Free Radic Biol Med 49(11): 1638-1645.
- Heinzelmann S, Bauer G (2010) Multiple protective functions of catalase against intercellular apoptosis-inducing ROS signaling of human tumor cells. Biol Chem 391(6): 675-693.
- Calvani M, Pelon F, Comito G, Taddei ML, Moretti S, et al. (2014) Norepinephrine promotes tumor microenvironment reactivity through β3-adrenoreceptors during melanoma progression. Oncotarget 6(7): 4615-4632.
- Dal Monte M, Casini G, Filippi L, Nicchia GP, Svelto M, et al. (2013) Functional involvement of β3-adrenergic receptors in melanoma growth and vascularization. J Mol Med (Berl) 91(12): 1407-1419.
- Bloom JD, Dutia MD, Johnson BD, Wissner A, Burns MG, et al. (1992) Disodium (R,R)-5-[2-[[2-(3-chlorophenyl)-2-hydroxyethyl]-amino] propyl]-1,3-benzodioxole-2,2-dicarboxylate (CL 316,243). A potent beta-adrenergic agonist virtually specific for beta 3 receptors. A promising antidiabetic and antiobesity agent. J Med Chem 272(28): 3081-3084.
- Atgiè C, D’Allaire F, Bukowiecki LJ (1997) Role of β1- and β3- adrenoceptors in the regulation of lipolysis and thermogenesis in rat brown adipocytes. Am J Physiol 273: C1136-C1142.
- Donadelli M, Dando I, Dalla Pozza E, Palmieri M (2015) Mitochondrial uncoupling protein 2 and pancreatic cancer: A new potential target therapy. World J gastroenterol 21(11): 3232-3238.
- Calvani M, Cavallini L, Tondo A, Spinelli V, Ricci L, et al. (2018) β3- adrenoreceptors control mitochondrial dormancy in melanoma and embryonic stem cells. Oxid Med and Cell Long p. 10.
- Yoshioka Y, Kadoi H, Yamamuro A, Ishimaru Y, Maeda S (2015) Noradrenaline increases intracellular glutathione in human astrocytoma U-251 MG cells by inducing glutamate-cysteine ligase protein via β3- adrenoceptor stimulation. Eur J Pharm 772: 51-61.
- Hadi T, Douhard R, Dias AM M, Wendremaire M, Pezzè M (2017) Beta3 adrenergic receptor stimulation in human macrophages inhibits NADPH oxidase activity and induces catalase expression via PPAR γ activation. Biochim Biophys Acta 1854(10): 1769-1784.
- Negre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, et al. (1997) A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J 11: 809-815.
- Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, et al. (2002) Superoxide activates mitochondrial uncoupling proteins. Nature 415: 96-99.
- Drisko JA, Chapman J, Hunter VJ (2003) The use of antioxidants with first-line chemotherapy in two cases of ovarian cancer. J Am Coll Nutr 22(2): 118-123.
