 Research Article
Research ArticleAbstract
Background: Managing relapsing metastatic, head and neck squamous cell carcinoma (HNSCC) is challenging. The genomic landscape of HNSCC has been recently described by numerous studies; however clinical utility studies are absent.
Methods: We describe next generation sequencing (NGS) results and clinical outcomes of eight patients with advanced recurrent HPV-negative HNSCC. NGS platform (FoundationOneTM) included the coding sequence of 236 cancer-related genes and 47 introns.
Results: All eight cases harbored ≥1 Genomic alterations (GA) accumulating to a total of 31 GA (average 3.9/tumor): the most common GA were; 10 in TP53 (6 tumors) and 5 in CDKN2A. Three GA were associated with FDA-approved drugs for HNSCC (EGFR amplification; Cetuximab), or other tumors (mTOR inhibitors - FBXW7-R465C; BRAF inhibitors - BRAF-K601E). Clinical trials associated with specific gene alterations were found to be applicable in all cases.
Conclusions: NGS in HNSCC is clinically feasible, revealing actionable GA in a high percentage of patients. Larger studies with access to early-phase clinical trials should improve outcome.
Keywords: Head and neck; Genomic sequence; P53; EGFR
Abbreviations: HNSCC: Head and Neck Squamous Cell Carcinoma; NGS: Next Generation Sequencing; GA: Genomic Alterations; CLIA: Clinical Laboratory Improvement Amendments; MVA: Modified Vaccinia Virus Ankara
Introduction
Managing advanced, recurrent, and metastatic head and neck cancer is challenging. Treatment options are limited, and prognosis is poor even when aggressive combinations of radiotherapy and chemotherapy are administered [1]. The addition of biological agents such as cetuximab prolongs survival by only a few months [2]. The inability to cure or even, in many cases, to slow the progression of the tumor has prompted investigators to search for potential new targets of systemic therapy. The introduction of nextgeneration sequencing has reduced the cost and processing time of genetic profiling, making it feasible in clinical scenarios [3,4]. However, although a myriad of genetic alterations can be detected with the available techniques, their translation into a benefit for patients with cancer presents a major hurdle [5]. The aim of this study was to explore genetic alterations in advanced, recurrent head and neck squamous cell carcinoma (HNSCC) using a commercially available next-generation sequencing platform. The literature was reviewed to evaluate the clinical implications of the findings.
Materials and Methods
Patients
The study group consisted of 8 patients with advanced recurrent HNSCC treated at a tertiary cancer center from 2012 to 2013. Their clinical characteristics are shown in Table 1. There were 4 men and 4 women of mean age 51.5 years at diagnosis (range 26-68 years). Tumor sites included the tongue (3 patients), skin/ parotid (2 patients), and larynx, nasopharynx, and tonsil (1 patient each). All tumors were negatively immunostained for p16 and were considered HPV negative. Five patients had advanced disease at presentation. Seven patients were surgically treated with curative intent, followed by radiotherapy and platinum-based chemotherapy. One patient (with nasopharyngeal cancer) underwent radiotherapy and chemotherapy only. The mean number of treatment lines before submission of the tumors for next-generation profiling was 3.4. Five patients (63%) died of their disease; mean time from nextgeneration sequencing to death was 11 months. The other three patients are alive with persistent disease after follow-up of 1, 10.5, and 27 months. Treatment decision-making was influenced by the sequencing results in one patient, as detailed below.
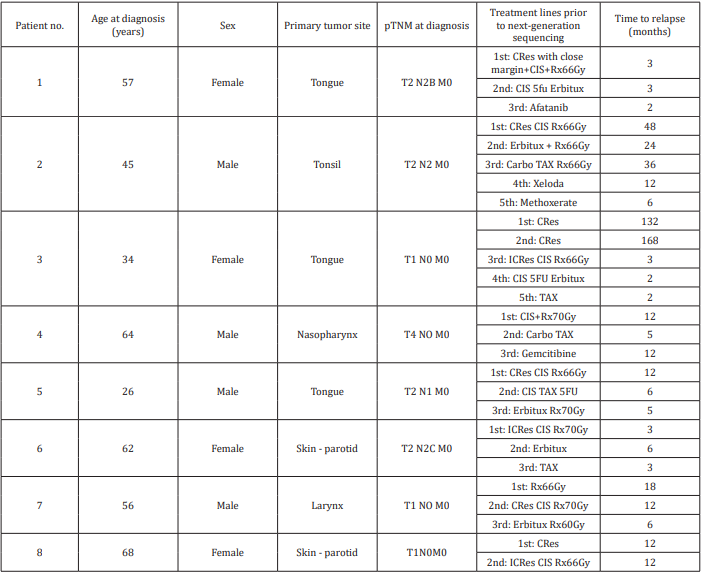
Table 1: Clinical characteristics of 8 patients prior to next-generation sequencing.
Next Generation Sequencing
Next-generation sequencing was performed with the Clinical Laboratory Improvement Amendments (CLIA)-approved FoundationOne™ platform (Foundation Medicine, Cambridge, MA, USA). FoundationOne is a targeted assay designed for use in routine formalin-fixed paraffin-embedded cancer specimens as small as 40μm. It simultaneously sequences the entire coding sequence of 236 cancer-related genes (3769 exons) plus 48 introns from 20 genes that are often rearranged or altered in cancer, to an average depth coverage of >250X. The assay detects all classes of genomic alterations: base substitutions, insertions and deletions, copy number alterations and rearrangements. A full list of sequenced cancer-related genes is available on line at the Foundation Medicine.
Results
Next-generation sequencing yielded 31 alterations in 19 genes, with a mean alteration/tumor ratio of 3.9 (range, 1-8 alterations per tumor). Two genes were predominantly involved: TP53 (10 mutations in 6 tumors) and CDKN2A (5 mutations in 5 tumors). One patient had a mutation only in TP53. There was one alteration in each of the following genes: CDKN2B, NOTCH1, EGFR, APC, CCND1, ARID1A, ARID2, SRC, FBXW7, SMARCA4, BRAF, ATM, MLH1, GRIN2A, ASXL1, MLL2 and NOTCH2. Three patients had mutations associated with FDA- approved treatments. In the first, genetic profiling identified EGFR amplification in a chest wall metastasis of a tongue carcinoma, along with mutations in APC-I1307K. Cetuximab has been approved for use in patients with HNSCC associated with EGFR amplification. However, it had already been administered according to standard protocol for 3 months prior to genetic analysis, with no response.
The second patient had a mutation in FBXW7-R465C and treatment with mTOR inhibitors such as everolimus and temsirolimus was suggested. These drugs have been FDA-approved for kidney, breast, and pancreatic cancers and reportedly yielded good results in 3 of 5 patients with HNSCC and either PIK3CA mutations or PTEN loss [6]. A combination of rapamycin (sirolimus) and vorinostat, an HDAC inhibitor that was suggested based on an additional alteration in the SMARCA4 gene, was administered. PETCT scan performed two months after treatment initiation revealed progressive disease in the neck and a new mediastinal lesion, and treatment was stopped. The third patient had metastatic cutaneous SCC. Next-generation sequencing of a parotid metastasis found a mutation in BRAF (K601E) and treatment with BRAF-inhibitor (vemurafenib) was considered. This treatment was avoided based on a history of cutaneous SCC and a reported increased risk for cutaneous SCC in melanoma patients treated with this drug [7]. On the basis of the FoundationOne assay thorough literature search, clinical trials were suggested in all cases based on their relevancy to 21 gene alterations (Table 2).
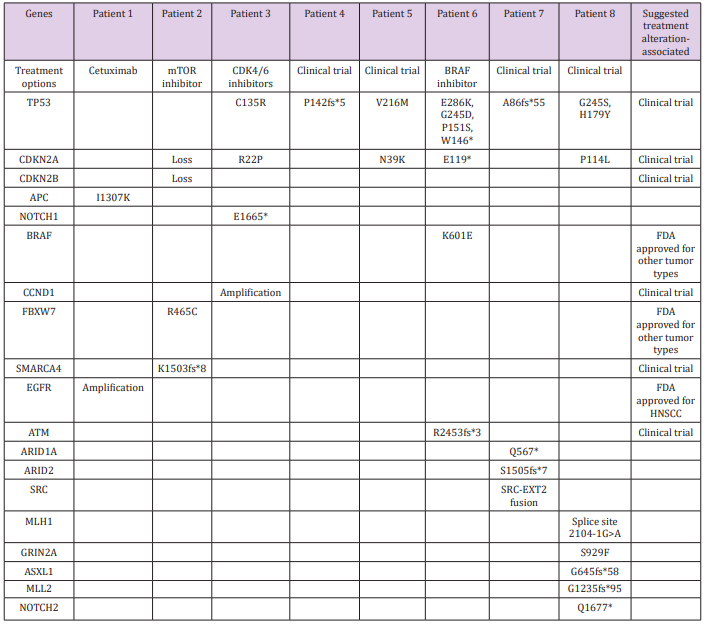
Table 2: Genetic alterations detected by next-generation sequencing.
Suggested treatments and clinical trials for actionable gene alterations are detailed in the right column.
Discussion
There is a standard first-line treatment protocol for recurrent, unresectable, metastatic HNSCC, but no successful second-line treatment. Next-generation sequencing is a novel tool that can help clinicians tailor treatment on the basis of gene alterations in tumors. Using the Foundation One platform, Drilon et al. [8] associated RETfusion positivity in non-small cell lung cancer with a good clinical response to the RET-inhibitor cabozanitib. Similarly, Vaishnavi et al. [9] associated a novel rearrangement in NTRK1-fusion with a clinical response to crizotinib. Prompted by these studies, we sought to explore the potential and utility of the next-generation sequencing approach to recurrent HNSCC. In the present study, profiling of 236 cancer-related genes in tumors from eight patients with advanced, recurrent, HNSCC yielded 31 genetic alterations (mean, 3.9 alterations per tumor). All tested tumors were affected.
Of the 19 genes with active alterations, only 2, TP53 and CDKN2A, had more than one mutation. Each mutation in both these genes was unique to the specific tumor. Comparable number of genetic alterations in HPV- negative HNSCC was described by Zhang P et al. [10] (3.2 per tumor). Genetic alterations associated with FDA- approved therapies were detected in 3 patients, while all other patients had actionable genetic alterations associated with treatments evaluated in clinical trials. Johnson et al. [11] assessed tumor from the head and neck as well other sites, with a similar platform. They found actionable alterations in 83%, of which 26% had a mutation associated with an FDA-approved drug for the specific site, and additional 17% had mutations associated with a drug approved for a different tumor type [11].
Potential Treatments for TP53 Altered Tumors
TP53 was altered in tumors from 6 of the 8 patients. Ten different mutations were found: 4 in one tumor, 2 in one tumor, and one mutation in each of the 4 remaining tumors. TP53 mutations are common in HNSCC as well as other types of cancer. Using the same sequencing platform, Chung et al. [12] demonstrated over 80% prevalence of TP53 mutations in HPV-negative HNSCC [12]. Similarly, Nichols et al. [13] identified TP53 mutations in 6/6 HNSCC cell lines [13], and Kandoth et al. [14] in 209/300 HNSCC tumors (70%) [14]. This rate is similar to our finding (75% of tumors had TP53 mutations) even though, the vast majority of tumors in that large series were primary (not recurrences).
There are currently no FDA-approved treatments directed against mutant p53. However, several treatment strategies have been evaluated: vaccines, gene therapy, and tyrosine kinases. The p53 synthetic long peptide (p53-SLP) vaccine targets cells that overexpress p53 and has been tested in patients with advanced colorectal [15] and ovarian [16] cancer. It induced a significant immunologic response in most patients and was found to be well tolerated [16,17]. However, clinical benefit could not be demonstrated. Another vaccine, the recombinant modified vaccinia virus Ankara (MVA) expressing wild-type human p53 (MVAp53), was found to induce p53-specific IFN-γ production, degranulation, cell proliferation, and lysis of p53-overexpressed tumor cells [18]. A phase II trial of two different p53 peptide vaccines showed specific immune responses with minimal toxicity in patients with ovarian cancer. The clinical efficacy of these vaccines was not assessed in this study and no control group was included [19].
A form of gene therapy, rAd-p53, delivers human recombinant wild-type p53 protein. It has had mixed results in phase II trials in hepatocellular carcinoma, ovarian cancer, and lung cancer [19-21]. Intraperitoneal injection of rAd-p53 in patients with heavily treated recurrent ovarian cancer decreased CA-125 levels in 50%. However, its injection to the hepatic artery in patients with hepatocellular carcinoma and to the bronchial artery in patients with nonsmall cell lung cancer yielded no benefit despite a significant immunologic response. In one study, intratumorally injections of Ad5CMV-p53, an adenoviral vector delivering wild type TP53, were administered to 10 patients with unresectable, chemoradiationresistant, esophageal squamous cell carcinoma. The treatment was found to be safe and well tolerated. Three patients showed absence of disease on repeated biopsies [22, 23].
MK-1775 is a potent and selective Wee-1 tyrosine kinase inhibitor. It targets CDC2 to inactivate the complex that regulates the G2 checkpoint. Studies in cell lines and animal models found that MK-1775 sensitizes p53- deficient tumor cells to chemotherapeutic agents and radiation; no sensitization was observed in p53 wildtype tumor cells [24,25].The suggestion that reactivation of p53 may serve as a potential treatment option led to the development of two alternative small-molecule approaches: stabilization of mutant p53 to rescue its DNA- binding activity, and inhibition of MDM2 or MDM4. The results are reviewed by Brown et al. [26] there are no clinical data currently supporting treatment with these agents.
Potential Treatments for CDKN2A-Altered Tumors
The CDKN2a gene was altered in 5 of our tumors. Studies using next-generation sequencing reported a CDKN2a mutation in 5/6 HNSCC cell lines [13], and 21% of primary HNSCC tumors in the largest series published [14].This rate is significantly lower than ours (63%; Fisher’s exact test, p=0.01), a difference that can suggest an increased aggressiveness of CDKN2A mutant tumors, or to a susceptibility of heavily treated tumors to acquire these mutations after the initial presentation. CDKN2A may be inactivated in HNSCC through deletion, promoter methylation, or mutation [27-30]. Tumors with loss of the CDKN2A/CDKN2B locus may be sensitive to CDK4/6 inhibitors, and clinical trials of these agents are currently underway. In preclinical melanoma studies, the response to CDK4 inhibition correlated with genomic changes that increase CDK4 activity, most notably where the tumor suppressor CDKN2A is deleted [31]. Similarly, using glioblastoma cells, researchers found that CDKN2A deletion predicts improved sensitivity to CDK4/6 inhibitors [32]. No CDK4/6 inhibitors are currently approved by the FDA for HNSCC.
FDA-Approved Treatments Suggested by Next-Generation Sequencing
Genetic alterations associated with FDA-approved therapies were detected in 3 patients in the present study (1 mutation each). One had an EGFR amplification, which has been reported in up to 27% of HNSCC cases [33]. EGFR encodes the epidermal growth factor receptor, a receptor tyrosine kinase that passes biochemical messages to the cell and stimulates it to grow and divide. Gene amplification, or mutation, or overexpression of EGFR protein may cause excessive proliferation and tumor formation [34]. Moreover, EGFR amplification and protein expression have been shown to be markers of poor prognosis in HNSCC [35]. Cetuximab is a monoclonal antibody that targets EGFR and was approved for treatment of HNSCC in a phase III trial. However, EGFR amplification did not predict response to this treatment [36]. Other EGFR-targeted therapies, including the tyrosine kinase inhibitors erlotinib, gefitinib, and lapatinib, have been approved by the FDA for use in other cancer types; in phase II trials, lapatinib and gefitinib induced a good response in cases of EGFR overexpression and amplification [37,38].
Currently, EGFR inhibition is well accepted in the treatment of HNSCC whether EGFR is overexpressed or not. Therefore, most patients will be given cetuximab prior to genetic evaluation, and a subsequent finding of EGFR amplification will not influence the treatment. Indeed, our patient had already been treated with cetuximab prior to genetic profiling. The drug was discontinued because of a poor response. FBXW7-R465C alteration is also associated with an FDA-approved drug, though not for HNSCC. It occurs at a mutation hot spot for this gene within the highly conserved WD40 repeat region. Mutations in this region may result in the failure to target substrates for degradation, thereby stabilizing the known oncogenic substrates Cyclin E, Notch1, MYC, Jun, and mTOR [39,40]. In an in vitro study, R465C disrupted recognition of its substrate (Cyclin E), resulting in substrate stabilization.
FBXW7 loss has been associated with increased cell proliferation and poor prognosis in esophageal squamous cell carcinoma [41]. In preclinical studies, FBXW7 inactivation stabilized the mTOR signaling protein and conferred sensitivity to rapamycin, an mTOR inhibitor. FBXW7 inactivation may also result in resistance to anti-tubulin chemotherapies [42]. NOTCH1 is one of the major targets of FBXW7 for degradation. Agrawal et al. [43] observed that FBXW7 mutations in HNSCC were in a hotspot known to block the degradation of active NOTCH1. In our study, one patient had a NOTCH1 E1665* mutation. As NOTCH1 can act as either an oncogene or tumor suppressor, depending on its cellular context, it may harbor both activating and inactivating mutations in cancer [44,45]. On the basis of its protein structure, E1665* is predicted to be an inactivating mutation. Genetic analysis of 74 HNSCCs revealed that 14% had mutations in NOTCH1 [46].
In contrast to acute T-cell lymphoblastic leukemia in which the activating NOTCH mutations are found in the heterodimerization or PEST (proline, glutamic acid, serine, threonine-rich) domain [47], in HNSCC, NOTCH1 mutations are largely dispersed elsewhere in the gene or produce truncated products. Therefore, NOTCH1 mutations in HNSCC are thought to represent loss-of-function changes. Activating mutations may be targeted via NOTCH inhibitors; however, there are currently no therapies available to address NOTCH1 inactivation. The BRAF V600E mutation is present in approximately 50% of metastatic melanoma cases, and vemurafenib, a BRAF inhibitor was approved by the FDA in these cases. Fifteen to 30% of patients treated with vemurafenib develop non-melanoma skin cancer; possibly due to a paradoxical activation of ras mutations in SCC that leads to accelerated growth of these lesions [7].
One tumor in our study presented a mutation in BRAF (K601E). Treatment with vemurafenib was avoided based on the history of cutaneous SCC in that patient. Most of the altered genes in our study are tumor suppressor genes: TP53, CDKN2A/B, APC, NOTCH1, FBXW7, ATM, ARID1a, ARID2. Similarly, Zhang P et al. [10] demonstrated that single nucleotide polymorphisms in tumor suppressors were more common in HPV- negative HNSCC in comparison with HPV positive tumors. Currently, treatment strategies directed at tumor suppressor genes are limited.
To conclude, Next-generation sequencing makes it possible to detect a diversity of gene alterations in patients with advanced, recurrent HNSCC who were heavily treated prior to genetic analysis. In the present study, the application of this technique identified actionable gene alterations in 6 of 8 patients (75%). The majority of alterations were not associated with FDA-approved treatments. In 3 cases, we had 6 treatment options. Two patients showed no objective response and 3 patients were referred for clinical trials. In 2 patients, only mutations in TP53 mutation were detected. Access to early-phase clinical trials may have improved the outcome of our cohort. Further studies are needed to improve understanding of the clinical utility of next- generation sequencing in HNSCC.
References
- Bernier J, Domenge C, Ozsahin M, Matuszewska K, Lefèbvre JL, et al (2004) Postoperative irradiation with or without concomitant chemotherapy for locally advanced head and neck cancer. N Engl J Med 350(19): 1945-1952.
- Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, et al. (2008) Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 359(11): 1116-1127.
- Shendure J, Ji H (2008) Next-generation DNA sequencing. Nat Biotechnol 26(10): 1135-1145.
- Reis Filho JS (2009) Next-generation sequencing. Breast Cancer Res 11(Suppl 3): S12.
- Natrajan R, Wilkerson P (2013) From integrative genomics to therapeutic targets. Cancer Res 73(12): 3483-3488.
- Holsinger FC, Piha Paul SA, Janku F, Hong DS, Atkins JT, et al. (2013) Biomarker-directed therapy of squamous carcinomas of the head and neck: targeting PI3K/PTEN/mTOR pathway. J Clin Oncol 31(9): e137- 140.
- Su F, Viros A, Milagre C, Trunzer K, Bollag G, et al. (2012) RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 366(3): 207-215.
- Drilon A, Wang L, Hasanovic A, Suehara Y, Lipson D, et al. (2013) Response to Cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 3(6): 630-635.
- Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, et al. (2013) Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med 19(11): 1469-1472.
- Zhang P, Neena M, Ada B, Fernandes H (2014) Molecular Heterogeneity of Head and Neck Squamous Cell Carcinoma Defined by Next-Generation Sequencing. The American Journal of Pathology 184(5): 1323-1330.
- Johnson DB, Kimberly H Knol J, Knol J, Gilbert J, Puzanov I, et al. (2014) Enabling a Genetically Informed Approach to Cancer Medicine: A Retrospective Evaluation of the Impact of Comprehensive Tumor Profiling Using a Targeted Next-Generation Sequencing Panel. The Oncologist 19(6): 616-622.
- Chung C H, Guthrie B, Masica D L, Tokheim C, Kang H, et al. (2015) Genomic alterations in head and neck squamous cell carcinoma determined by cancer gene-targeted sequencing. Annals of Oncology 26(6): 1216-1223.
- Nichols AC, Yoo J, Palma DA, Fung K, Franklin JH, et al. (2012) Frequent mutations in TP53 and CDKN2A found by next-generation sequencing of head and neck cancer cell lines. Arch Otolaryngol Head Neck Surg 138(8): 732-739.
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, et al. (2013) Mutational landscape and significance across 12 major cancer types. Nature 502(7471): 333-339.
- Speetjens FM, Kuppen PJ, Welters MJ, Essahsah F, Voet van den Brink AM, et al. (2009) Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin Cancer Res 15(3): 1086-1095.
- Leffers N, Lambeck AJ, Gooden MJ, Hoogeboom BN, Wolf R, et al. (2009) Immunization with a P53 synthetic long peptide vaccine induces P53- specific immune responses in ovarian cancer patients, a phase II trial. Int J Cancer 125(9): 2104-2113.
- Leffers N, Vermeij R, Hoogeboom BN, Schulze UR, Wolf R, et al. (2012) Long-term clinical and immunological effects of p53-SLP® vaccine in patients with ovarian cancer. Int J Cancer 130(1): 105-112.
- Song GY, Srivastava T, Ishizaki H, Lacey SF, Diamond DJ, et al. (2011) Recombinant modified vaccinia virus ankara (MVA) expressing wildtype human p53 induces specific antitumor CTL expansion. Cancer Invest 29(8): 501-510.
- Rahma OE, Ashtar E, Czystowska M, Szajnik ME, Wieckowski E, et al. (2012) A gynecologic oncology group phase II trial of two p53 peptide vaccine approaches: subcutaneous injection and intravenous pulsed dendritic cells in high recurrence risk ovarian cancer patients. Cancer Immunol Immunother 61(3): 373-384.
- Buller RE, Runnebaum IB, Karlan BY, Horowitz JA, Shahin M, et al. (2002) A phase I/II trial of rAd/p53 (SCH 58500) gene replacement in recurrent ovarian cancer. Cancer Gene Ther 9(7): 553-566.
- Tian G, Liu J, Zhou JS, Chen W (2009) Multiple hepatic arterial injections of recombinant adenovirus p53 and 5-fluorouracil after transcatheter arterial chemoembolization for unresectable hepatocellular carcinoma: a pilot phase II trial. Anticancer Drugs 20(5): 389-395.
- Guan YS, Liu Y, Zou Q, He Q, La Z, et al. (2009) Adenovirus-mediated wildtype p53 gene transfer in combination with bronchial arterial infusion for treatment of advanced non-small-cell lung cancer, one-year followup. J Zhejiang Univ Sci B 10(5): 331-340.
- Shimada H, Matsubara H, Shiratori T, Shimizu T, Miyazaki S, et al. (2006) Phase I/II adenoviral p53 gene therapy for chemoradiation resistant advanced esophageal squamous cell carcinoma. Cancer Sci 97(6): 554- 561.
- Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, et al. (2010) MK1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther 9(7): 514-522.
- Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, et al. (2011) MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res 17(17): 5638-5648.
- Brown CJ, Cheok CF, Verma CS, Lane DP (2011) Reactivation of p53: from peptides to small molecules. Trends Pharmacol Sci 32(1): 53-62.
- Reed AL, Califano J, Cairns P, Westra WH, Jones RM, et al. (1996) High frequency of p16 (CDKN2/MTS-1/INK4A) inactivation in head and neck squamous cell carcinoma. Cancer Res 56(16): 3630-3633.
- Worsham MJ, Pals G, Schouten JP, Van Spaendonk RM, Concus A, et al. (2003) Delineating genetic pathways of disease progression in head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg 129(7): 702-708
- Worsham MJ, Chen KM, Tiwari N, Pals G, Schouten JP, et al. (2006) Finemapping loss of gene architecture at the CDKN2B (p15INK4b), CDKN2A (p14ARF, p16INK4a), and MTAP genes in head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg 132(4): 409-415.
- Wong TS, Man MW, Lam AK, Wei WI, Kwong YL, et al. (2003) The study of p16 and p15 gene methylation in head and neck squamous cell carcinoma and their quantitative evaluation in plasma by real-time PCR. Eur J Cancer 39(13): 1881-1887.
- Sheppard KE, McArthur GA (2013) The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin Cancer Res 19(19): 5320-5328.
- Wiedemeyer WR, Dunn IF, Quayle SN, Zhang J, Chheda MG, et al. (2010) Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc Natl Acad Sci U S A 107(25): 11501- 11506.
- Chau NG, Perez Ordonez B, Zhang K, Pham NA, Ho J, et al. (2011) The association between EGFR variant III, HPV, p16, c-MET, EGFR gene copy number and response to EGFR inhibitors in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Head Neck Oncol 3: 11.
- Ciardiello F, Tortora G (2008) EGFR antagonists in cancer treatment. N Engl J Med 358(11): 1160-1174.
- Chang SS, Califano J (2008) Current status of biomarkers in head and neck cancer. J Surg Oncol 97(8): 640-643.
- Licitra L, Mesia R, Rivera F, Remenár E, Hitt R, et al. (2011) Evaluation of EGFR gene copy number as a predictive biomarker for the efficacy of cetuximab in combination with chemotherapy in the first-line treatment of recurrent and/or metastatic squamous cell carcinoma of the head and neck: EXTREME study. Ann Oncol 22(5): 1078-1087.
- Del Campo JM, Hitt R, Sebastian P, Carracedo C, Lokanatha D, et al. (2011) Effects of lapatinib monotherapy: results of a randomised phase II study in therapy-naive patients with locally advanced squamous cell carcinoma of the head and neck. Br J Cancer 105(5): 618-627.
- Saarilahti K, Bono P, Kajanti M, Bäck L, Leivo I, et al. (2010) Phase II prospective trial of gefitinib given concurrently with cisplatin and radiotherapy in patients with locally advanced head and neck cancer. J Otolaryngol Head Neck Surg 39(3): 269-276.
- Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, et al. (2007) FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res 67(19): 9006-9012.
- Mao JH, Kim IJ, Wu D, Climent J, Kang HC, et al. (2008) FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 321(5895): 1499-1502.
- Yokobori T, Mimori K, Iwatsuki M, Ishii H, Tanaka F, et al. (2012) Copy number loss of FBXW7 is related to gene expression and poor prognosis in esophageal squamous cell carcinoma. Int J Oncol 41(1): 253-259.
- Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, et al. (2011) Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471(7336): 110-114.
- Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, et al. (2011) Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 333(6046): 1154- 1157.
- Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez Irizarry C, et al. (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306(5694): 269-271.
- Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, et al. (2011) Loss-offunction mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U S A 108(43): 17761-17766.
- Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, et al. (2011) The mutational landscape of head and neck squamous cell carcinoma. Science 333(6046): 1157-1160.
- Lee SY, Kumano K, Masuda S, Hangaishi A, Takita J, et al. (2005) Mutations of the Notch1 gene in T-cell acute lymphoblastic leukemia: analysis in adults and children. Leukemia 19(10): 1841-1843.
