 Research Article
Research ArticleAbstract
First-principles calculations are performed on the dissociation of H2 molecule and subsequent spillover of hydrogen atoms over Al(111) surface doped by two Ti. Two catalytic activity configurations are formed by replacement of the nearest neighbor or the next nearest neighbor Al atoms of the Al(111) surface with two Ti atoms, respectively. Calculations show that both dopes sites are energetically favorable. Compared with on the Ti@Al(111) surface, the hydrogen dissociation barrier of H2 molecule will decrease 0.40 eV and 0.29 eV, respectively. The diffusion barrier height of one hydrogen atom on the 2Ti@Al(111) surface is moderate, 0.59 eV and 0.66 eV for one possible paths, 0.63eV, 0.52eV, 0.61eV, 0.50 eV for another possible paths. Our results potentially lead to dope many other effective catalytic elements to improve the reversible in the reaction of hydrides.
Keywords: First-Principles Calculations; Al(111) Surface; Dissociation and Diffusion; Barrier
Introduction
The catalytic activation of hydrogen gas is an important step involved in many applications, for example, fuel cells, hydrogenation, petroleum refining, especially, one of intense interest issues hydrogen storage, where hydrogen gas is as an energy carrier. Various hydrogen storage materials [1-5] have been investigated, and the most promising materials are complex metal hydrides, which are cheap, safe, reliability and with high storage capacity (3-12 wt%), but they have many problems, like slow kinetics of hydrogen uptake and release, high decomposition temperature and nearly irreversible. Sodium alanate (NaAlH4 ) is one of such materials. The release and uptake of hydrogen can be summarized by two-step in NaAlH4 [6-9]:
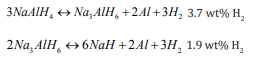
In 1997, Bogdanović and schwickardi [1] proposed doping a few amounts of Ti-containing material (β-TiCl3 ) to NaAlH4 , which was a major breakthrough, for it could accelerate the reaction rates of both directions, reduce the decomposition temperature, most importantly rehydrogenate the products into the initial material, meeting all the needing criteria of on board applications. Moreover, Ti was a special catalyst for NaAlH4 , which had little impact on the hydrogen reversibility compared with many other transition metals.
Of course, an ideal catalysis of H2 molecule should make the dissociation easy, the binding of intermediates and substrate weak, and especially abundant on the earth. Therefore, dissociation of H2 molecule on pure or doped aluminum surface is surging. Chaudhuri et al reported that the energy barrier of hydrogen atom diffusion from a catalytic hollow site to an adjacent hollow site was 1.57 eV on two Ti atoms next nearest neighbor doped Al(001) [10]. Du obtained the dissociation barriers for first and second H2 molecules on Ti atom doped Al(001) surface were 0.21 and 0.235 eV, respectively, where the diffusion barrier was 0.47 eV [11]. However, H2 dissociation was quite high on pure Al(111) with the barrier 1.28 eV. When doped one Ti atom on the surface and subsurface, the dissociation barrier decreased by 0.62 eV and 0.57 eV, with diffusion barriers, 0.90 eV and 0.42 eV, respectively [11]. In 2011, the catalytic activity of two Ti atoms doped on Al(111) surfaces was clear showed by Chopra using CO2 as a detector [12], for the formed complexes were metastable with different desorption temperatures. Mahboob obtained that H2 dissociation and spillover on the Al(111) surface was effected by the placement of Ti atom [13]. Based on these finding, Here we investigate the dissociation and diffusion of H2 on the two Ti atom doped Al(111).
Computational Methods
First principles calculations are performed in the Vienna Ab Initio Simulation Package (VASP) code [14,15]. We employ the Projector Augmented Plane Wave (PAW) method with PerdewWang generalized gradient approximation [16,17]. The two Ti atoms doped Al(111) surfaces is constructed using 4×4×4supercell, with four atomic layers. A vacuum distance of 15Å is chosen to avoid interactions between images. In calculation, the downmost Aluminium atomic layer is fixed while other three layers are fully relaxed. The sampling of the brillouin zone is represented by (7×7×1) grid meshes. An energy cutoff of 340eV is chosen. All of the geometric structures are relaxed until the energy and the force less than 1×10-4eV and 0.01 eV/Å, respectively.
To determine the dissociation and diffusion energy barrier of H2 molecule on the 2Ti@Al(111) surfaces, Configuration Interaction Nudged Elastic Band (cNEB) method is used to find the Minimum Energy Paths (MEP), where 4 to 20 images are constructed between given initial and final state. The elastic connected images are optimal with a damped molecular dynamics. The force is set to 0.03 eV/Å.
Results and Discussion
In experiment, we have already known that when two Ti atoms doped in the Al(111) surface, there exists two catalytic activity sites, the nearest neighbor and next nearest neighbor configurations, where the corresponding distances of Ti atoms are 2.9Å and 4.9Å, respectively. There is no doubt the stability of 2Ti@Al(111) surface. In theory, the formation energy is calculated by
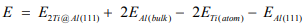
Where E2Ti@Al(111) and EAl(111) are the total energy of two Ti atoms doped and pure Al(111) surface, ETi(atom) and EAl(bulk) are the energy of a single Ti atom and a bulk Al atom. Under this definition, when two Ti atoms are nearest neighbor, the total formation energy is -11.73eV. The average formation energy of one Ti atom can be -5.87 eV. The corresponding values of next nearest neighbor configuration are -11.94eV and -5.97eV, respectively. This indicates the latter is more stable, about 0.1eV/Ti atom, for negative formation energy means the doped surface with lower energy. The reason is maybe, it will impede dope the other Ti atom after one Ti atom is doped in the Al(111) surface.
Before the barrier calculations, the binding energy of H atom adsorbed in pure Al(111) surface is studies. There are four specific adsorption sites (top, bridge, fcc hollow and hcp hollow). The preferable adsorption site is fcc hollow, meaning the lower dissociative barrier. However, the four sites are with a little of energy difference, similar to the results of Wang [11]. To seeking the barriers, we calculate all the possible sites of two Ti atoms doped on the Al(111) surface.
First, the H2 molecule is put 6Å above and parallel to the two Ti atoms nearest neighbor doped 2Ti@Al(111) surface. Then, the model is optimized and approached step by step. When the distance between them is 1.9 Å, the H2 molecule adsorbs in the surface, with adsorption energy -0.43 eV and H–H bond 0.82Å. Compared to the free hydrogen gas molecule (0.74 Å), the bond is elongated about 0.08 Å. This state is identified as the initial state (IS) of H2 molecule dissociation. The Final State (FS1) is chosen from the lowest energy configuration, i.e., fcc-hcp sites between two Ti atoms, as shown in Figure 1 (left). The energy pathway is created by inserting eight elastic connected images between IS and FS1. Though optimization, we can obtain MEP and the dissociation barrier of H2 molecule 0.26 eV. Associated with the previous case, the barrier decrease 1.02 eV and 0.40 eV than on the pure Al(111) surface and one Ti doped in the surface [11]. Repeating the same process for H2 molecules on next nearest neighbor configuration, the IS determined by the H2 molecule with a distance of 1.98Å from the surface. Meanwhile, the adsorption energy is -0.41eV and the elongation of H-H bond is 0.07Å. The FS1 is hydrogen atoms in bridge-bridge sites, as shown in Figure 1 (right). Eight images are built between IS and FS1. In the NEB calculation, we find 0.37eV of hydrogen dissociation barrier, 0.11eV higher than the barrier of nearest neighbor configuration.
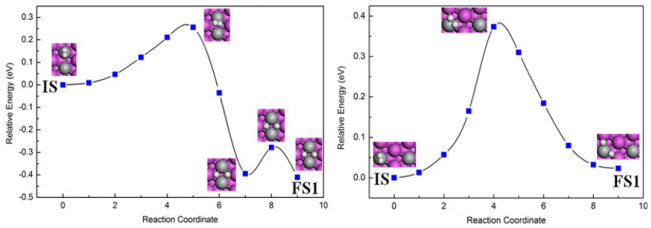
Figure 1: The dissociation of H2 molecule on the two Ti atoms doped Al(111) surfaces, nearest neighbor configuration (left) and next nearest neighbor configuration (right).
Further, the diffusion barrier of one hydrogen atom migration is calculated. The relaxed FS2 is found by comparing the energies of models with one or both hydrogen atoms move a step away from the catalytic site. Two kinds of FS2 are picked out, which both are bridge-bridge sites as inset images of Figure 2, with the energy difference of 0.06 eV. Twenty images are used to linked to FS1 and two kinds of FS2, respectively, which are eight, four and eight images from FS1 to Mid to FS2. The diffusion barrier heights are also moderate 0.59 eV (black line) and 0.66 eV (red line). The results show it is favorable to hydrogen atom diffusion than on one Ti atom doped Al(111) surface, which is 0.9 eV [11].
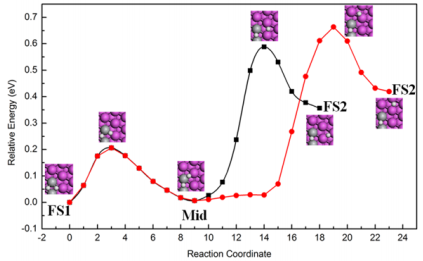
Figure 2: The spillover of one dissociation hydrogen atom on the two Ti atoms nearest neighbor doped Al(111) surface.
For the two Ti atoms next nearest neighbor doped configuration, the diffusion is complex. The FS is four different bridge-bridge sites, demonstrated in Figure 3a-3d, which the energy differences are not more than 0.035 eV, with four possible diffusion paths. Between the activation site and final migration site, we insert sixteen images. The corresponding barriers are 0.63eV (black line), 0.52eV (red line), 0.61eV (blue line), 0.50eV (green line). Therefore, the diffusion of one hydrogen atom away from dissociation site on the next nearest neighbor model is relatively easier than on the nearest neighbor model.

Figure 3: The spillover of one dissociation hydrogen atom on the two Ti atoms next nearest neighbor doped Al(111) surface, green line (a), blue line (b), red line (c), black line (d).
Conclusion
On the 2Ti@Al(111) surface, the catalytic activation of H2 molecule is investigated via ab initio DFT calculations. The process is divided into two steps, dissociation and migration away from the catalytically active site. There are formed two catalytic activity configurations when the nearest neighbor or next nearest neighbor surface Al atoms are substituted by Ti atoms. Both configurations are favorable to dissociation H2 molecule with the barrier heights of 0.26 eV and 0.37 eV, respectively, which is decreased about 0.40 eV and 0.29 eV in comparison with on the Ti@Al(111). Then, one hydrogen atom is migration away from the Ti site. There are two possible diffusion paths for the nearest neighbor 2Ti@Al(111) surfaces with the barriers of 0.59 eV and 0.66 eV, respectively, and four possible diffusion paths for the next nearest neighbor with the barriers of 0.63eV, 0.52eV, 0.61eV, and 0.50 eV, respectively. The barriers are moderate for the barrier height reaches 0.90 eV on Ti@Al(111) surface. Our results indicate the dissociation of H2 molecule and subsequent diffusion are particularly favorable on 2Ti@Al(111) surface. The existence of two catalytic activity sites observed in experiment can be well explained. The high catalytic activity of next nearest-neighbor 2Ti@Al(111) surface combine lower diffusion barriers and more diffusion paths, so the dissociation hydrogen atoms can migrate immediately. The low dissociation barrier of H2 molecules on the nearest neighbor configuration, while the higher diffusion barriers and less diffusion paths make hydrogen atoms together leading to the reduction of the catalytic activity.
Acknowledgement
This work is supported by the PhD Starting Fund of Guangdong Ocean University (120702/R17077), by the Innovation and Strong School Research Project of Guangdong Ocean University (No. GDOU2017052621).
References
- Bogdanovic B, Schwickardi M (1997) Ti-doped alkali metal aluminium hydrides as potential novel reversible 1 hydrogen storage materials. J Alloys Compd 253: 1-9.
- Schlapbach L, Zuttel A (2001) Hydrogen-storage materials for mobile applications. Nature 414: 353-358.
- Eigen N, Keller C, Dornheim M, Klassen T, Bormann R (2007) Industrial production of light metal hydrides for hydrogen storage. Scr Mater 56: 847-851.
- Eberle U, Felderhoff M, Schüth F (2009) Chemical and physical solutions for hydrogen storage. Angew Chem Int Ed 48: 6608-6630.
- Soler L, Macanás J, Muñoz M, Casado J (2007) Synergistic hydrogen generation from aluminum, aluminum alloys and sodium borohydride in aqueous solutions. Int J Hydrog Energy 32: 4702-4710.
- Bogdanovic B, Felderhoff M, Kaskel S, Pommerin A, Schlichte K, et al. (2003) Improved hydrogen storage properties of Ti-doped sodium alanate using titanium nanoparticles as doping agents. Adv Mater 15: 1012-1015.
- Anton DL (2003) Hydrogen desorption kinetics in transition metal modified NaAlH4 . J Alloys Compd 356: 400-404.
- Gross KJ, Sandrock G, Thomas GJ (2002) Dynamic in situ X-ray diffraction of catalyzed alanates. J Alloys Compd 330: 691-695.
- Jensen CM, Gross KJ (2001) Development of catalytically enhanced sodium aluminum hydride as a hydrogen-storage material. Appl Phys A 72: 213-219.
- Du AJ, Smith SC, Lu GQ (2007) The catalytic role of an isolated-Ti atom in the hydrogenation of Ti-doped Al(001) surface: An ab initio density functional theory calculation. Chem Phys Lett 450: 80-85.
- Wang J, Du Y, Kong Y, Xu H, Jiang J, et al. (2010) The effect of Ti atom on hydrogenation of Al(111) surface: First-principles studies. Int J Hydrog Energy 35: 609-613.
- Chopra IS, Chaudhuri S, Veyan JF, Chabal YJ (2001) Turning aluminium into a noble-metal-like catalyst for low-temperature activation of molecular hydrogen. Nat Mater 10: 884-889.
- Al-Mahboob A, Muller E, Karim A, Muckerman JT, Ciobanu CV, et al. (2012) Site-dependent activity of atomic Ti catalysts in al-based hydrogen storage materials. J Am Chem Soc 134: 10381-10384.
- Kresse G, Furthmuller J (1996) Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci 6: 15-50.
- Kresse G, Furthmuller J (1996) Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B 54: 11169-11186.
- Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77: 3865-3868.
- Kresse G, Joubert D (1999) From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B 59: 1758-1775.
