 Research Article
Research ArticleAbstract
The manufacture of some gene chips used for high throughput analyzing methylation status is based on the construction CpG islands. By now the reported method used to construct CpG islands database is loaded down with trivial details and is difficult to screen effective clones. In this study we combined suppression subtraction hybridization with methylation to develop a simple method used to construct CpG islands database. In the method the driver DNAs were prepared using linker-PCR that the genomic DNA fragments of Chinese fir made by restriction enzyme were linked with linkers and then cut with the methylation-sensitive restriction enzyme HapII and that as template the linker-PCR were completed. The genomic DNA treated with Methylase Sss I were as the tester and after linked with adaptors the tester were cut by HapII.
After two hybridizations and two suppressive PCR, the PCR products were cloned into vectors and the detection of fragments inserted into vectors was performed with PCR and dot hybridization method then sequencing. It was found that among the randomly 536 clones there were 517 clones that had an insert that was more than 100bp long. In 175 selected clones, 159 clones were positive clones, and 81.7% clones were identified as CGI clones according the sequencing analysis. All these results proved the method shows great potential in constructing CpG islands database in Chinese fir.
Keywords: CpG Islands Database; Suppression Subtraction Hybridization; Methylation; Chinese Fir
Introduction
Chinese fir (Cunninghamia lanceolate (Lamb.) Hook) is the most important conifer species for commercial wood production in southern China Zheng et al. [1]. Chinese fir accounts for about 25 % of total plantation area in China and about 50 % of domestic plantation timber production in southern China. Now, the breeding cycle for Chinese fir get into third generation Pan [2]. However, most research about Chinese fir was forced on traditional breeding Zhang, et al. [3]. The study of molecular biology was still not completely clear. DNA methylation profiling had become an important technology in many areas of epigenetics research Zhang, Kimatu et al. [4]. Methylation profiling approaches were aimed at investigating DNA methylation patterns either across the entire genome and at different levels of resolution, or they were used to study methylation patterns at specific areas of interest, for example at promoters and other regulatory elements Fatemi et al. [5]. In genetics, CpG islands were genomic regions that contain a high frequency of CpG sites but to date objective definitions for CpG islands were limited. The usual formal definition of a CpG island was a region with at least 200 bp and with a GC percentage that was greater than 50% and with an observed/expected CpG ratio that was greater than 60% Gardiner Garden et al. [6].
At present, a variety of methods were used to evaluate the methylation status of genes: southern blot Bickle et al. [7], bisulfite genomic DNA sequencing Frommer et al. [8], restriction enzymePCR Kane et al. [9], methylation-specific PCR (MSP) Frommer et al. [8], methylation-sensitive single nucleotide primer extension (MSSnuPE) Herman et al. [10], electrochemistry Kuppuswamy et al. [11] and so on. However, each method was restricted to the evaluation of DNA methylation on gene-by-gene basis. Such an approach had given researchers a limited picture of complex epigenetic alterations in plants. Therefore, a compelling need existed to develop novel technologies for the next phase of epi-genome research. A powerful approach for studying the genetic nature of many biological processes was to characterize genes that vary in expression level during this process. Suppression Subtractive hybridization (SSH) Diatchenko et al. [12,13], was a technology that allowed for PCRbased Von Stein et al. [14-16] amplification of only cDNA fragments that differ between a control and experimental transcriptome. Differences in relative abundance of transcripts were highlighted, as were genetic differences between species. The technique relied on the removal of dsDNA formed by hybridization between a control and test sample, thus eliminating DNA or gDNA of similar abundance, and retaining differentially expressed, or variable in sequence, transcripts or genomic sequences. The purpose of this study was to explore the feasibility of enrichment methylation CpG islands of genome through combining the suppression subtraction hybridization with methylation in Chinese fir.
Materials and Methods
DNA Extraction and Enzyme Treatment
The materials were obtained from Yangkou Forestry Center (Fujian, China). Genomic DNA was isolated by standard methods using CTAB method Gu et al. [17]. The DNA derived from the buds of Chinese fir was divided into two parts. One aliquot was treated by methylase SssI (SM) as positive control with the conditions recommended by the supplier (New England Biolabs, Beverly, MA). The other was not treated by methylase SssI as negative control. The positive control generated in this way had 100% methylated cytosine in the test CpG sites, whereas the negative control had all unmethylated cytosine residues in the test CpG sites. Approximately 2–3 ug genomic DNA was restricted to completion with 50 U of MseI following the conditions recommended by the supplier (New England Biolabs). This enzyme restricted bulk DNA into small fragments. As its recognition site (TTAA) rarely occurred in GC-rich regions, most CpG islands remain intact after the restriction. The digests were purified with QIAquick column (QIAGEN).
Driver Preparing
The cleaved ends of DNA were ligated to unphosphorylated linkers. The linker sequences were H-24 (5’-AGG CAA CTG TGC TAT CCG AGG GAT-3’), and H-12 (5’-TAATCC CTC GGA -3’) Hou et al. [1]. The use of universal linkers allowed to subsequently amplifying all methylated fragments in ligated DNA samples. The ligation step began with the annealing of H-12 to H-24 (each 100uM) by mixing equal amounts of each oligonucleotides in a microcentrifuge tube and allowing the mixture to cool from 55 ℃ to room temperature over 1 hour. The annealed linker-primers (10ul/sample) were then added to the purified DNA, together with 2.0ul 10×T4 DNA Ligase Buffer, 2.0ul 10mM ATP, 1 ul T4 DNA Ligase (Takara), and water to 20ul. The ligation reaction was carried out at 16℃ overnight. The mixture was then held 65 ℃ for 10 min to stop the reaction. The ligated products were purified with QIAquick column (QIAGEN) and digested with the methylation-sensitive endonucleoase HapⅡ following the conditions recommended by the supplier (New England Biolabs).
The digest was used for PCR without further purification. PCR reactions were performed for only 20 cycles. At this level of amplification, especially if most unmethylated repetitive sequences were restricted away by the HapⅡ digestion, the proportions of PCR products for most templates were expected to be in the linear range. PCR reactions were performed in a 50ul volume, containing 0.4 uM H-24 primer; 4 U Taq DNA polymerase (Takara); 5% DMSO; 200uM dNTP; 2.5mM Mg2+ in a buffer provided by the supplier. The tubes were incubated for 3 min at 72 ℃ to fill in 5’-protruding ends of the ligated DNA and subjected to 20 cycles of amplification consisting of 1 min at 95℃ and 3 min at 70 ℃. The final extension was lengthened to 10 min
Tester Preparing
Adaptor1 (10uM) and 2 (Figure 1) were each separately ligated to 2ul of 1:6 dilution of SM-digested DNA (Tester) at 16℃ overnight. Samples were then heated at 72℃ for 5 minutes to inactivate. Then extracted with phenol chloroform and resuspended in 5.5μl H2O and digested with the HapⅡ.
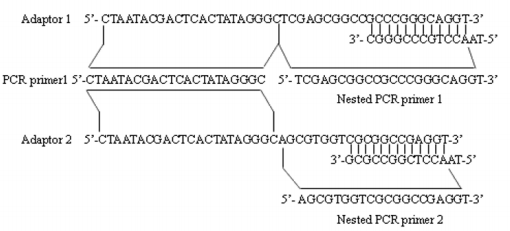
Figure 1: Sequences of adaptors and adaptor primers used in MS-SSH. The illustration shows the detailed sequences of adaptors (i.e., adaptor 1 and 2) and adaptor primers (i.e., PCR primer 1 used in primary PCR, Nested PCR primer 1 and 2 used in secondary PCR) used in the MS-SSH system.
Two Subtractive Hybridization
In the first hybridization, 1.5μl HapⅡ-digested normal DNA (Driver) was mixed with 1.5μl of diluted adaptor1-ligated or adaptor 2-ligated SM -digested DNA (Tester) (about 15 copies of Driver DNA vs. 1 copy of Tester) in the presence of hybridization buffer. The samples were denatured at 98℃ for 1.5 min and immediately incubated in 68℃ for 6-8 hours. In the second hybridization, the two samples from the first hybridization were mixed in the presence of a freshly denatured (1μl of 1:2 dilution) Driver DNA. The sample was incubated at 68 ℃ for 12 hours. After adding 200μl of dilution buffer, the sample was incubated for an additional 7 minutes.
Two Suppression PCR (Nested PCR)
The primary polymerase chain reaction (PCR) was conducted in 20μl volume containing 1μl of the diluted subtraction mixture, 1× PCR reaction buffer, 10uM PCR primer1 (Figure 1), 0.2mM dNTPs and U Taq DNA polymerase (Takara). The reaction mixture was incubated at 75 ℃ for 5 minutes to extend the adaptors, followed by 94℃ for 2 minutes , then followed by 35 cycles at 94℃ for 30 seconds, 66℃ for 30 seconds, and 72℃ for 1.5 minutes. The primary PCR mixture was diluted 20 fold, and 1μl was used in secondary PCR with Nested primers for primers 1 and 2 (Figure 1). The conditions of the reaction were 94℃ for 30 seconds , 66℃ for 45 seconds , and 72℃ for 1.5 minutes for 30 cycles.
Construction of 5’CpG Islands Fragments Libraries
The products of the secondary PCR were verified by agarose gel electrophoresis (AxyPrep DNA Gel Extraction Kit), and then cloned into the pMD 19-T Vector (Takara, Dalian). The detection of recombinant clones was based on blue/white selection. All recombinant clones were picked and culture overnight in 3mL of LB medium containing 100mg/L ampicillin in 10mL centrifuge tube at 37 ℃. The inserts were amplified by PCR using Nested PCR primer 1 and 2. The PCR products were electrophoresed on a 1% agarose gel to confirm the amplification quality and quantity
Dot-Blot Hybridization
To further confirm positive clones, the inserts were amplified by PCR using the Nested primer 1 and 2; the PCR products were purified by Takara DNA Fragment Purification Kit Ver.2.0, and then dotted into Hybond-N+ nylon membranes (Amersham, Piscataway, New Jersy). The probes (MS-digested DNA and undigested DNA) were labeled by digoxigenin (DIG). Hybridization and detection were carried out following the DIG High Prime DNA Labeling Detection Kit protocol.
Sequencing of Bands
The positive clones were sequenced with vector primers. The Advanced BlastN and BlastX programs at the NCBI website (http:// www.ncbi.nlm.nih.gov/) were respectively used for homology analysis of the cloned DNA sequences that gave quality-reads Benson et al. [18,19].
Result
MS–SSH was an efficient way to isolate and enrich genomic CpG islands. The driver DNA was prepared by Linker–PCR combined with Mse I and methylation-sensitive restriction endonuclease (i.e., Hpa II) digestion of genomic DNA, and then the SSH procedure was performed to isolate and enrich methylated CpG islands contained in tester DNA. The amplicons were generated by the procedures outlined above (Figure 2).Firstly, The size distribution of MseIdigested genomic DNA ranged from 100 to 2000bp (Figure 3B). Secondary, the MseI-digested DNA was ligated to H-24/H-12 linkers combined with subsequent digestion of HpaII to prepare MseI–HpaII pretreated DNA by Linker–PCR amplifications (Figure 3D). Genomic fragments containing methylated sites were protected from the methylation-sensitive enzymes digestion and could be amplified by linker-PCR. Many of these linker-ligated MseI fragments were expected to be present in the sample due to aberrant methylation in the test sites, whereas the same unmethylated fragments were digested and not present in the normal application after PCR. Finally, the MseI–HpaII pretreated amplicons were prepared by MseI digestion of their corresponding products of Linker–PCR to remove the H24/H12 linkers at the ends of DNA molecules (Figure 3E).
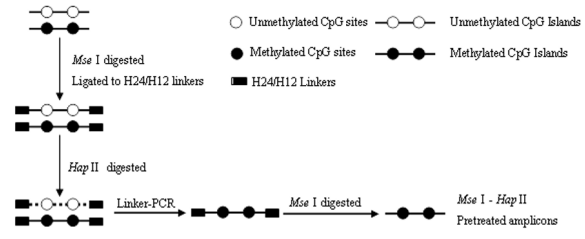
Figure 2: Schematic flowchart of driver generation.
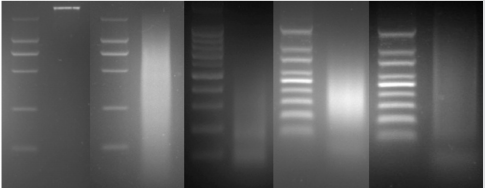
Figure 3: Amplicon generation of MS–SSH system. (A) Genomic DNA. (B) MseI digestion of genomic DNA. (C) MseI–HpaII pretreated DNA. (D) Linker–PCR products of MseI–HpaII pretreated DNA. (E) MseI digestion of the Linker–PCR products. White triangles indicate the H-24/H-12 linkers cleaved from both ends of the DNA molecules.
Isolation and Construction of CpG Islands
Firstly, the secondary PCR products of hybridization were evaluated by agarose gel electrophoresis (Figure 4A). Because there was no tester-specific DNA fragments contained in tester DNA, one visible smear mainly in the 200 to 1000bp range could be seen in the secondary PCR products of subtraction. After the tertiary PCR products of MS-SSH subtraction were purified, they were cloned into the pMD–19 T vector to prepare the CpG Islands library. To estimate the proportion of CpG Islands-like clones in the CpG Islands library, the size distribution of cloned inserts was firstly evaluated by PCR amplification of insert fragments using primers (i.e., Nested primer 1 and 2) that was flanking the cloned inserts. Altogether, 536 clones were randomly selected and analyzed, with 96.5% (517/536) of cloned inserts ranging from 200 to 1000bp and the remainder being longer than 1000bp (Figure 4B). The resulting insert fragments were ranged from 200 to 1000bp (Figure 4B). Finally, recombinant plasmids were digested by EcoR I and Hind III to verify the positive recombinants. For the above 536 clones, PCR fragments of 192 cloned inserts were further digested with EcoR I and Hind III, with 91.1% (175/192) containing one insert (Figure 4C). Thus, a putative subtracted MS-SSH library of Chinese fir was constructed.
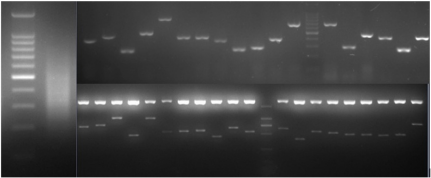
Figure 4: Characteristics of the cloned inserts in the CGI library. (A) Secondary PCR products. (B) Representative amplified cloned inserts from the CGI library. (C) EcoR I/Hind III double digest of recombinant plasmids.
Differential Screening of MS-SSH Library
To characterize the differential gene expression of the identified 175 CpG Islands clones, a dot blot was performed, as both DNA subtraction and differential screening procedures utilized PCR amplified material. Several control DNAs were also utilized to validate this technique. Only 16 false-positive clones were detected and the rest were confirmed as ‘‘true’’ CpG Islands (Figure 5). As a more thorough evaluation of the CpG Islands library, the insert sequences of 159 randomly selected clones were determined by DNA sequencing. Subsequently, further sequence analyses were performed, including restriction site analysis, CpG Island Searcher (http://cpgislands.usc.edu/) to analyze the base composition (GC%) and frequency of observed CpG versus expected CpG (ObsCpG/ExpCpG) (Figure 6), and NCBI BLAST to search for homologous sequences in the GenBank database (Figure 7A). The results showed that 90.9% (145/159) containing inserts having one or more HpaII sites. In the 145 clones, bulk DNA (GC% < 40%, ObsCpG/ExpCpG < 0.6) was contained in 15 clones (9.4%, 15/159). The last 130 (81.7%, 130/159) were identified as CpG Islands clones because they satisfied the minimal criteria for CpG Islands length > 200 bp, GC% > 50%, ObsCpG/ExpCpG > 0.6), of which only 15 (10.3%, 15/145) were duplicated clones. All insert fragments in the 123 CpG Islands clones were single copy in genomic DNA based on the results of NCBI BLAST homology searching (Figure 7B).

Figure 5: Differential screening of clones from the identified CpG Islands clones. Quadruplicate colony PCR products were prepared and the membrane was hybridized with DIG-labeled DNA probers derived from (a) tester DNA pool and (b) driver DNA pool. T.C., tester control; D.C., driver control. Arrows indicate false-positive clones.
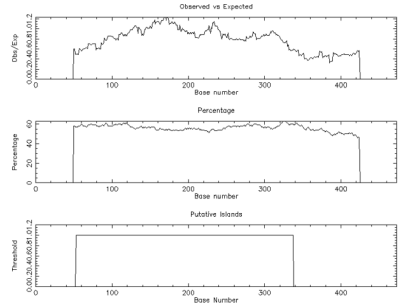
Figure 6: The result of CpG Detetion.
Note: CpGPLOT islands of unusual CG composion: EMBOSS-001 from 1 to 474 (Observed/Expected ratio>0.60; Percent C+percent G>50.00 and length>200.).
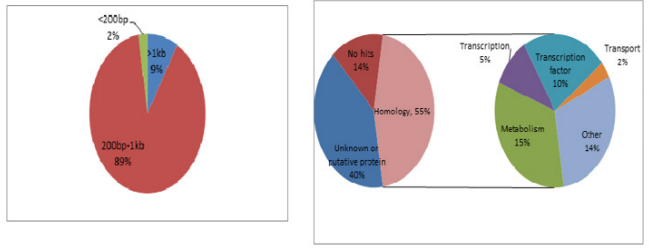
Figure 7: Characteristics of the cloned inserts in the CpG Islands library. (A) Size distribution of inserts of 159 randomly selected clones. (B) The results of NCBI BLAST homology searching.
Sequences Analysis of MS-SSH Library
Of the 69 aligned sequences, there are 19 genes with high similarity to known metabolism in GeneBank (Table 1), including oxidases, dehydrogenases, phosphatases, protein kinases, N-acetylglucosaminyltransferases, and proteins of unknown function. Among them, the MF1701 fragment has high homology with the pentatricopeptide repeat (PPR) protein, which is an RNA specific binding protein, and play important roles in plant growth and development, organelle formation, fertility restoration of cytoplasmic male sterility, RNA Editing and processing, signal transduction between nuclear and organelles, stress defense and so on. Table 1 Sequence of methylated fragments and database search.
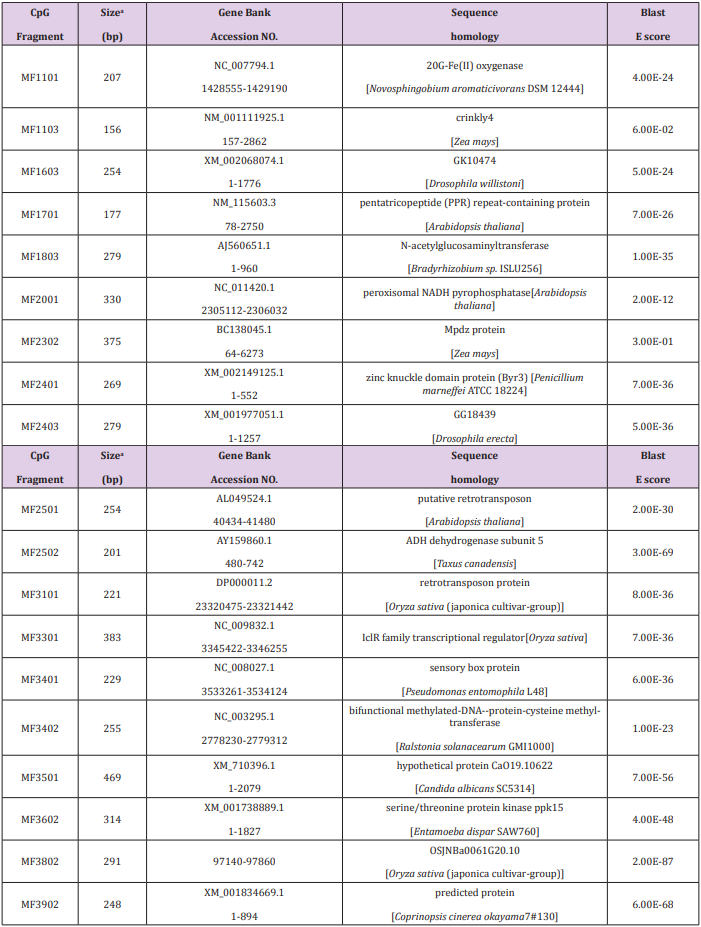
Table 1: Sequence of methylated fragments and database search.
Discussion
DNA methylation was referred to as epigenetic phenomena that control various genomic functions without a change in nucleotide sequence Morris [20]. DNA methylation was associated with early embryo of plant organizations development Suzuki et al. [21], genetic imprinting Razin et al. [22], and chromosome inactivation Chan et al. [23]. Therefore, the detection method of DNA methylation was rapid development. Among various DNA methylation analysis methods, chip techniques have shown powerful abilities to isolate and identify the aberrant methylated CGIs. Such methods include MCA-RDA (Methylation in CpG island amplification-representational difference analysis) Toyota et al. [24], DMH (Differential methylation hybridization using CGI array) Yan et al. [25], MSO (Methylation specific oligonucleotide) Yu et al. [26], ChIP-on-Chip (Chromatin immunoprecipitation) Kuras [27]. Subtractive hybridization was a powerful technique that enables researchers to compare two populations of DNA and obtain clones of genes that were expressed in one population but not in the others Diatchenko et al. [13]. Although there were several different methods, the basic theory behind subtraction was simple.
First, we refer to the one DNA that contains specific CpG Islands as tester, and the reference DNA as driver. In this research, tester and driver DNA were hybridized, and the hybrid sequences were then removed. Consequently, the remaining unhybridized CpG Islands that were expressed in the tester yet absent from the driver (Figure 4). In some cases, the MS-SSH method greatly enriches for differential CpG islands; nevertheless, the subtracted sample would still contain some templates that correspond to CpG islands common to both the tester and driver samples Illingworth et al. [28]. Although this background may depend somewhat on the quality of DNA purification and performance of the particular subtraction, in chiefly arises when DNA species were differentially expressed in tester and driver. In general, a limited set of differentially expressed DNA and low quantitative difference in expression produces higher background, even if the sufficient enrichment of differentially expressed DNAs were obtained Ogino et al. [29]. With high background, picking random clones from the CpG Islands library for Southern blot analysis was extremely time-consuming and inefficient.
Therefore, differential screening should be performed before embarking on Southern blot analysis. In this study, it was appeared some un-CpG Islands, which may be related to the design of experiment (Figure 5). After M.Sss I enzyme treatment, all CpG sites of genomic DNA were methylated, and could not digested by HapⅡ in the tester. Nevertheless, the unmethylated CGIs in the driver were digested by HapⅡ. Therefore, the discrepancy fragments between tester and driver could amplify through the suppression PCR. Cytosine methylation influences gene expression, a hypothesis based on the frequency of methylcytosine and on the proposed transmission of methylation patterns through cycles of DNA replication Finnega et al. [30-32]. The quality of CpG islands library was close relationship with the following experiment. Therefore, it is very importance for testing the quality of CpG islands library through HTF-HapⅡ tiny fragment analysis of tools-Sequence Analysis-CpG plot software in European Bioinformatics Institute (EBI). The construction of library in the research is very high of CpG content, also can be used to detect new genes and following experiment (Figure 6).
Conclusion
Development of the MS-SSH technology not only significance for the CpG islands database construction, also advanced for high-throughput comparative analysis the difference between the organizations methylation.
Acknowledgement
This study was supported by the Program for Fujian Province Science and Technology Research funding on the fourth Tree Breeding Cycle Program of Chinese fir (Grant No. Min Lin Ke 2016- 35). We were grateful to all researcher of gene laboratory Room 60537 in the Key Laboratory of Forest Genetics and Biotechnology at Nanjing Forestry University for constructive suggestions to improve the manuscript.
References
- Zheng R, H Zhou, S Su, L Bian, X Hui, J Shi, and H. X. Wu (2016) Inheritance of growth and survival in two 9-year-old, open-pollinated progenies of an advanced breeding population of Chinese firs in southeastern China. Journal of Forestry Research 27(5): 1-9.
- Pan L (2017) Contrastive Analysis on Early Growth among Chinese Fir 3~(rd) Generation Seed Orchard. Fujian Forestry.
- Zhang Y, X Han, J Sang, X He, M Liu, et al. (2016) Transcriptome analysis of immature xylem in the Chinese fir at different developmental phases. Peerj 4(17): e2097.
- Zhang M, JN Kimatu, K Xu, B Liu (2010) DNA cytosine methylation in plant development. Journal of Genetics and Genomics 37(1): 1-12.
- Fatemi M, MM Pao, S Jeong, EN Gal Yam, G Egger, et al. (2005) Footprinting of mammalian promoters: use of a CpG DNA methyltransferase revealing nucleosome positions at a single molecule level. Nucleic acids research 33(20): e176-e176.
- Gardiner Garden M, M Frommer (1987) CpG islands in vertebrate genomes. Journal of molecular biology 196(2): 261-282.
- Bickle TA, D Krüger (1993) Biology of DNA restriction. Microbiological reviews 57(2): 434-450.
- Frommer M, LE Mc Donald, DS Millar, CM Collis, F Watt, et al. (1992) A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proceedings of the National Academy of Sciences 89(5): 1827-1831.
- Kane MF, M Loda, GM Gaida, J Lipman, R Mishra, et al. (1997) Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer research 57(5): 808-811.
- Herman JG, JR Graff, S Myöhänen, BD Nelkin, SB Baylin (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proceedings of the National Academy of Sciences 93(18): 9821-9826.
- Kuppuswamy MN, J Hoffmann, CK Kasper, SG Spitzer, SL Groce, et al. (1991) Single nucleotide primer extension to detect genetic diseases: experimental application to hemophilia B (factor IX) and cystic fibrosis genes. Proceedings of the National Academy of Sciences 88(4): 1143- 1147.
- Diatchenko L, Y Lau, AP Campbell, A Chenchik, F Moqadam, et al. (1996) Suppression subtractive hybridization: a method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proceedings of the National Academy of Sciences 93(12): 6025-6030.
- Gurskaya NG, L Diatchenko, A Chenchik, PD Siebert, GL Khaspekov, et al. (1996) Equalizing cDNA subtraction based on selective suppression of polymerase chain reaction: cloning of Jurkat cell transcripts induced by phytohemaglutinin and phorbol 12-myristate 13-acetate. Analytical biochemistry 240(1): 90-97.
- Von Stein, OD, WG Thies, M Hofmann (1997) A high throughput screening for rarely transcribed differentially expressed genes. Nucleic Acids Research 25(13): 2598-2602.
- Yamagishi H, V Garg, R Matsuoka, T Thomas, D Srivastava (1999) A molecular pathway revealing a genetic basis for human cardiac and craniofacial defects. Science 283(5405): 1158-1161.
- Yokomizo T, T Izumi, K Chang, Y Takuwa, T Shimizu (1997) A G-proteincoupled receptor for leukotriene B4 that mediates chemotaxis. Nature 387(6633): 620-624.
- Gu H, L Hu (1998) Laboratory handbook of plant molecular biology. Publishing House of High Education, Beijing, China 4-11.
- Benson DA, I Karsch Mizrachi, DJ Lipman, J Ostell, BA Rapp, et al. (2000) GenBank. Nucleic acids research 28(1): 15-18.
- Ye J, S Mc Ginnis, TL Madden (2006) BLAST: improvements for better sequence analysis. Nucleic acids research 34(2): W6-W9.
- Morris J (2001) Genes, genetics, and epigenetics: a correspondence. Science 293(5532): 1103-1105.
- Suzuki MM, A Bird (2008) DNA methylation landscapes: provocative insights from epigenomics. Nature Reviews Genetics 9(6): 465-476.
- Razin A, H Cedar (1994) DNA methylation and genomic imprinting. Cell 77(4): 473-476.
- Chan SWL, IR Henderson, SE Jacobsen (2005) Gardening the genome: DNA methylation in Arabidopsis thaliana. Nature Reviews Genetics 6(5): 351-360.
- Toyota M, C Ho, N Ahuja, KW Jair, Q Li, et al. (1999) Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer research 59(10): 2307-2312.
- Yan PS, CM Chen, H Shi, F Rahmatpanah, SH Wei, et al. (2001) Dissecting complex epigenetic alterations in breast cancer using CpG island microarrays. Cancer Research 61(23): 8375-8380.
- Yu YP, S Paranjpe, J Nelson, S Finkelstein, B Ren, et al. (2004) High throughput screening of methylation status of genes in prostate cancer using oligonucleotide methylation array. Carcinogenesis 26(2): 471-479.
- Kuras L (2004) Characterization of protein-DNA association in vivo by chromatin immunoprecipitation. Signal Transduction Protocols, Springer 147-162.
- Illingworth, R., A. Kerr, D. DeSousa, H. Jørgensen, P. Ellis, J. Stalker, D. Jackson, C. Clee, R. Plumb and J. Rogers (2008). A novel CpG island set identifies tissue-specific methylation at developmental gene loci.
- Ogino S, M Cantor, T Kawasaki, M Brahmandam, GJ Kirkner, et al. (2006). CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut 55(7): 1000-1006.
- Finnega EJ, RK Genger, WJ Peacock, ES Dennis (1998) DNA Methylation in Plants. Annu Rev Plant Physiol. Plant Mol. Biol. 49: 14.
- Bian Liming, ShiJisen, Zheng Renhua, Chen Jinhui, X Wuharry (2014) Genetic parameters and genotype–environment interactions of Chinese fi. Canadian Journal of Forest Research 44(6): 582-592.
- Hou P, M Ji, C Ge, J Shen, S Li, N He, Z Lu (2003) Detection of methylation of human p16Ink4a gene 5′‐CpG islands by electrochemical method coupled with linker‐PCR. Nucleic acids research 31(16): e92-e92.
