 Research Article
Research Article Abstract
Growth hormone pituitary adenomas (GHomas) are the second most common of pituitary tumors that hypersecrete prolactin. In clinically, the treatment and diagnosis of GHomas are limited, and the earlier and more frequent recurrences were demonstrated in invasive pituitary adenomasa. The identification of the association of genetic mutation and GHomas progression can provide significant interest to patients with GHomas. In recent years, studies of GHomas focused on the identification of genetic mutation in sporadic pituitary adenomas. Previously, a study has shown DPCR1, EGFL7, the PRDM family and LRRC5 in pituitary adenomas are probably functional in modifiers of tumorigenesis, development of oncocytic change and invasive tumor phenotype. The detection of genetic mutations still needs further study. Whole-exome sequencing, for spectrum of genetic alterations at present study, was performed on 7 sporadic GHomas DNA and corresponding blood samples. In total, 293 variants were predicted to be functionally damaged in 7 tumor samples (median 41 mutations/sample; range: 40-73).
4 candidate driver mutations, including RBM43, KRTAP4-9, GNGT2, and CENPW, were identified and functional significance analyses suggest that GNGT2, but not the other three genes, may contribute to the pathogenesis of GHomas. Lastly, gene ontology and pathway analyses identified mutations in 31 genes that were associated with metabolic pathways (22 genes), calcium signaling (7 genes) and adipocytokine signaling pathways (5 genes), respectively, implicating these pathways in the pathogenesis of GHomas. In conclusion, several candidate somatic mutations and changes in copy numbers for GHomas were identified. (The results showed no recurrence of mutations in the tumors studied but a low number of mutations, thereby highlighting their benign nature). Further studies on a larger cohort of TSHomas, along with the use of epigenetic and transcriptomic approaches, may reveal the underlying genetic lesions.
Conclusion: Our results demonstrated no recurrence of mutations in the tumors studied. Interestingly, the data suggested that metabolic, calcium signaling or adipocytokine signaling pathways might be involved in tumor pathogenesis, suggesting further studies on a larger cohort of tumor tissues. Further, epigenetic approaches may be beneficial in revealing the etiology of sporadic GH-secreting pituitary adenomas.
Keywords: Exome Sequencing; Sporadic GH-Secreting Pituitary Adenomas; Hub genes
Introduction
The incidence of pituitary adenomas in the population is approximately 1: 1000 [1], which is 15% of all intracranial neoplasms [2]. Growth hormone adenoma is an extremely common functional pituitary adenoma, second only to pituitary tumors that hypersecrete prolactin. Only 5% of pituitary adenomas occur in patients with a familial history [1] and most are sporadic Growth Hormone (GH)-secreting pituitary adenomas [3], which cause acromegaly in adults, and excessive GH secretion during childhood/ adolescence can lead to gigantism [4]. Pituitary adenomas are thought to be monoclonal in origin [5], but the genesis of pituitary tumors has remained controversial. These tumors can cause serious complications, including ophthalmological, neurological and endocrinological abnormalities. Currently, factors that increase tumor progression remain uncharacterized, although many of the genetic changes associated with familial pituitary adenomas, including Multiple Endocrine Neoplasia type 1 (MEN1), Carney’s complex and MEN4 [6-8], as well as germline mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene and X-chromosomal microduplication, can predispose individuals to pituitary adenomas [6,9].
Somatic mutations in GNAS have been identified in 30–40% of GH-producing pituitary adenomas [7]. Interestingly, 53% of Japanese patients with GH-secreting pituitary adenomas have been reported to exhibit somatic GNAS mutations [8]. Mutations in the gene that encodes the α subunit of stimulatory G-protein, Gs, are the only mutational changes unequivocally associated with GH-secreting adenomas [10]. Pathological analyses have identified proliferation markers that were unaltered in mutated GNAS pituitary tumors and non-mutated tumors, suggesting that the GNAS1 mutation affected secretion rather than proliferation [11]. Only 5% of pituitary adenoma patients have a familial history and despite multiple extensive studies, no oncogene or tumor suppressor genes have been discovered to directly affect the pathogenesis of sporadic GH-secreting pituitary adenomas. In order to discover genes involved in these adenomas, whole-exosome sequencing can be utilized to search for new somatic mutations in sporadic GH-secreting pituitary adenomas. Recently, Ronchi et al. examined adenoma tissue genomes by targeted sequencing (n = 31) and next-generation exome-sequencing (n = 36). No recurrent somatic mutations were observed, except for known alterations in the GNAS gene, which was like results collected by the wholegenome sequencing (n = 12) performed by Valimaki et al. [13,14]. Together, this indicates that pathogenesis in sporadic GH-secreting pituitary adenomas remains to be clarified. In order to further explore the somatic landscape of sporadic GH-secreting pituitary adenomas, we performed whole-exome sequencing coupled to rigorous analytical methods, which further confirmed novel recurrent genetic alterations and important mutated genes.
Materials and methods
Patients and Tumors
Seven patients (3 males and 4 females, mean age 42 years [range 20–77 years]) with sporadic GH-secreting pituitary adenomas, who did not have a family history of endocrine neoplasia (Table 1). The tumor samples were collected between 2012 and 2013 at the Shenzhen Second People’s Hospital and frozen while fresh. Tumor contents of adenomas were verified with hematoxylin and eosin stainings.
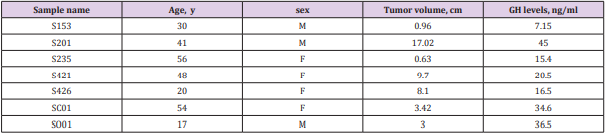
Table 1: Clinical Details of Patients.
Whole-Exome Sequencing and Bioinformatics Analysis
Exome enrichment was performed using the Agilent Sure Select Human All Exon V5+ UTR, while sequencing was performed with the Illumina Hiseq 4000 platform outputting 150-bp reads. Sequencing data were aligned to hg19/GRCh37 using the burrows-wheel aligner (bwa v0.7.12). Single nucleotide variants and deletions were identified using mutect-1.1.7 and strelka-1.0.11. Candidate driver mutations were identified using MutSigCV-1.4.
Gene Ontology and Pathway Enrichment Analysis for Mutated Genes
Gene Ontology analysis (GO) is a common method for annotating genes and gene products, while also for identifying characteristic biological attributes for high-throughput genome or transcriptome data. Kyoto Encyclopedia of Genes and Genomes (KEGG; http:// www.genome.jp) is a database that can be utilized in systematically analyzing gene functions and linking genomic information with higher-order functional information. Comprehensive mapping of a user’s gene to relevant biological annotations in the DAVID database (https://david.ncifcrf.gov) is essential for success in high-throughput gene functional analysis. In order to analyze the mutated genes at the functional level, GO enrichment and KEGG pathway analysis were performed in conjunction with the DAVID online tool. p < 0.05 was statistically significant.
Integration of Protein–Protein Interaction (PPI) Network and Module Analysis
The Search Tool for the Retrieval of Interacting Genes (STRING) database is an online tool designed to evaluate PPI information. STRING (version 9.0) covers 5,214,234 proteins from 1,133 organisms. In order to evaluate the interactive relationships among mutated genes, the genes were mapped to STRING, and only experimentally validated interactions with a combined >0.4 score were significant. PPI networks were then constructed using the Cytoscape software and the plug-in Molecular Complex Detection was used to screen the modules of the PPI network in Cytoscape. The criteria were set as follows: Molecular Complex Detection scores >3 and number of nodes >4. Function and pathway enrichment analyses were also conducted for mutated genes in the modules. p < 0.05 was statistically significant.
Results
Whole-Exome Sequencing
All tissues and blood samples of sporadic GHomas patients, matched by the exome analysis, were collected while informed consent was obtained. Whole-Exome sequencing, for spectrum of genetic alteration, was performed on 7 set of sporadic GHomas DNA and corresponding blood samples to identify a total of 2,197 high quality SNVs and tumor-specific deletions (313.86/tumor) in 7 GHomas (259, 238, 213, 424, 409, 266, 388, respectively) (Figure 1). From which, 293 variants were predicted to be functionally damaging, including 272 missense, 53 splice-site, and 13 nonsense variants. The entire list of somatic mutations was presented in Table 2. A total of 2197 high quality SNVs and tumor-specific deletions (313.86/tumor) were identified via whole-exosome sequencing of GH-secreting pituitary adenomas (Table 2 and Figure 1). 293 variants were predicted to be functionally damaging, including 272 missense, 8 splice-site and 12 nonsense variants. The entire list of somatic mutations is reported in Supplementary Table S1.
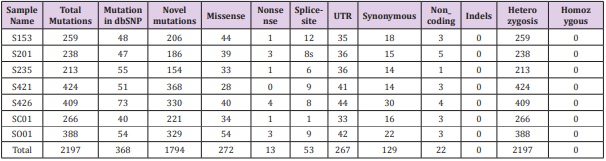
Table 2: Overview of somatic DNA mutations detected in 7 growth hormone adenoma samples.
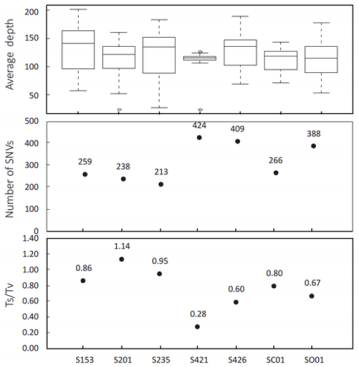
Figure 1: Statistics of high quality (minimum 10x coverage and 10% variant base read percentage) somatic SNVs reads: (i) The average sequencing depth of the target region within each chromosome, (ii) The number of somatic SNVs and (iii) The transition (Ts)– transversion (Tv) ratio of each tumor.
Identification of Candidate Driver Mutations
Consistent with the absence of a family history for pituitary adenomas, the tumor samples lacked germline mutations in genes encoding MEN1, CDKN1B, AIP, and PRKAR1A, which are familial genes in pituitary adenomas. In addition, no mutations were found in known oncogenes, tumor suppressor genes, or genes previously implicated in other pituitary tumors. In order to identify candidate driver mutations, MutSigCV-1.4 was used to correct for variations by using patient-specific mutation frequency and spectrum, as well as gene-specific background mutation rates that were incorporated into expression level and replication time [15]. Using these criteria, 4 candidate driver mutations genes were identified: RBM43, KRTAP4-9, GNGT2 and CENPW. Previously, only GNGT2 had been implicated in tumorigenesis and epigenetic changes.
Gene Ontology and Pathway Analyses
Using GO analyses, genes were significantly enriched in metabolic processes and biological regulation (Biological Processes), the gene products were concentrated in the membrane and nucleus (Cellular Component), and the functions related to protein and ion binding (Molecular Function) (Figure 2). Using the KEGG database pathway analysis tool, there were no significant overlaps, however the discovered genes were associated with metabolic, calcium signaling, and adipocytokine signaling pathways (Supplementary Figures S1-S4), Yet there was not significant enrichment of the pathways and, integrating the data with the PPI network and module analysis, none of the genes were recognized as hub-genes.
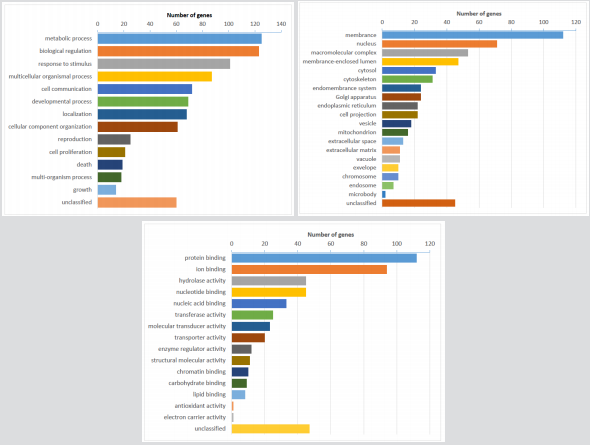
Figure 2: Gene ontology analysis of somatic mutations. (A) GO biological process classification; (B) GO cellular component classification; (C) GO molecular function classification.
Discussion
Whole-exome sequencing of sporadic GH-secreting pituitary adenoma tissues identified several somatic variants. The mutation rate (~41 mutations/tumor) was slightly higher than previously reported in GH-secreting pituitary adenomas (n = 12), GH-secreting pituitary adenomas (n = 36) and non-functioning (n = 7) pituitary adenomas (13, 14, 16), However, unlike previous studies of other pituitary adenomas, there were no mutations involving the MEN1, AIP, PRKAR1A, CDKN1B and GPR101, GNAS, GPR101, or USP8 genes [6,13,14,17,18]. 4 candidate driver mutations were observed in this study: RBM43, KRTAP4-9, GNGT2, and CENPW. Only GNGT2 variants have been associated with any diseases. GNGT2, localized to chromosome 17q21, encodes the guanine nucleotide binding protein, Gamma Transducing Activity Polypeptide 2 (GNGT2) [19], which is thought to play a crucial role in cone phototransduction.
However, GNGT2 is important in obesity, interacting with the chemokine signaling pathway to affect the risk of pancreatic cancer [20] and methylation of GNGT2 is linked to the tobacco smoking to risk of coronary artery disease [21]. The role of GNGT2 methylation in pituitary adenoma tumorigenesis and progression are needed to examine. Previous studies of pituitary adenoma have suggested that pituitary growth hormone imbalance is not caused by somatic mutation, but instead by epigenetic deregulation, including cytosine methylation, modification of histone proteins, or microRNA expression. Previous studies have reported that methylation plays a causative role in pituitary tumorigenesis [22] and both cell cycle regulators and putative tumor suppressor genes are methylated in pituitary adenomas [23-25]. For example, hypermethylation of the Rb gene promoter causes low expression levels of Rb in pituitary adenomas [26].
IK6 is a dominant-negative isoform of the transcription factor Ikaros, a zinc-finger DNA-binding protein, that is present in pituitary adenomas and has been shown to be epigenetically regulated through histone and DNA modifications [27,28]. Palmieri et al. observed reduced expression of multiple micro RNAs that target high mobility group A transcripts, HMGA1 and HMGA2, in each of the major pituitary adenoma subtypes [12]. Further studies are needed to examine the functional relevance of this pathway. Intriguingly, The GO term analysis showed that mutations genes were mainly involved developmental biological processes and ion transport in cancer cells differs substantially from normal cells [29]. Furthermore, the enriched KEGG pathways of mutations genes included calcium signaling, metabolic and adipocytokine signaling pathways, which indicates that these pathways are related, although there was no significant enrichment of these pathways.
Activated Ca2+ signaling leads to an increase in cytosolic free calcium, which then further triggers GH secretion [30,31]. These findings agree with a recent study that described whole-genome alterations in 12 GH-secreting tissues using next-generation exomesequencing (n = 36) [13,14]. Other pathways that participate in pituitary adenomas have not been reported, although some heterogeneous tumors have been observed to share a few common mutations [14]. Together, these data indicate that different genes act through the same molecular pathways and may contribute to tumor formation in sporadic GH-secreting pituitary adenomas.
Conclusion
We did not identify any novel or recurrent mutated genes from GH-secreting pituitary adenomas. However, these genetic findings, coupled with previous studies, suggests that different genes act through similar pathways or epigenetic changes, which may contribute to tumor occurrence and development.
Funding Source
This project was supported by the national natural science foundation of China (81772685), the international cooperation research projects of Shenzhen Science and Technology Program (GJHZ20160301163419476 and GJHZ20160301163900284).
References
- Beckers, A Aaltonen, LA Daly, AF Karhu A (2013) Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev 34(2): 239-277.
- Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, et al. (2006) A High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab 91(12): 4769-4775.
- Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, et al. (2004) The prevalence of pituitary adenomas: a systematic review. Cancer 101(3): 613-619.
- Sabino, SM Miranda, PA Ribeiro Oliveira A (2010) Growth hormonesecreting pituitary adenomas: from molecular basis to treatment options in acromegaly. Cancer Biol Ther 9(7): 483-492.
- Herman V, Fagin J, Gonsky R, Kovacs K, Melmed S (1990) Clonal origin of pituitary adenomas. J Clin Endocrinol Metab 71(6): 1427-1433.
- Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, et al. (2014) Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med 371(25): 2363-2374.
- Landis CA, Harsh G, Lyons J, Davis RL, McCormick F, et al. (1990) Clinical characteristics of acromegalic patients whose pituitary tumors contain mutant Gs protein. J Clin Endocrinol Metab 71(6): 1416-1420.
- Fukuoka H, Takahashi Y (2014) The role of genetic and epigenetic changes in pituitary tumorigenesis. Neurol Med Chir 54(12): 943-957.
- Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, et al. (2006) Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 312(5777): 1228-1230.
- Lania A, Spada A (2009) G-protein and signalling in pituitary tumours. Horm Res 71: 95-100.
- Freda PU, Chung WK, Matsuoka N, Walsh JE, Kanibir MN, et al. (2007) Analysis of GNAS mutations in 60 growth hormone secreting pituitary tumors: correlation with clinical and pathological characteristics and surgical outcome based on highly sensitive GH and IGF-I criteria for remission. Pituitary 10(3): 275-282.
- Beckers A, Aaltonen L, Daly AF, Auli Karhu (2013) Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocrine Reviews 34(2): 239-277.
- Ronchi CL, Peverelli E, Herterich S, Weigand I, Mantovani G, et al. (2016) Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur J Endocrinol 174(3): 363-372.
- Välimäki N, Demir H, Pitkänen E, Kaasinen E, Karppinen A, et al. (2015) Whole-Genome Sequencing of Growth Hormone (GH)-Secreting Pituitary Adenomas. J Clin Endocrinol Metab 100(10): 3918-3927.
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, et al. (2013) Mutational heterogeneity in cancer and the search for new cancerassociated genes. Nature 499(7457): 214-218.
- Newey PJ, Nesbit MA, Rimmer AJ, Head RA, Gorvin CM, et al. (2013) Whole-exome sequencing studies of nonfunctioning pituitary adenomas. J Clin Endocrinol Metab 98(4): 796-800.
- Vallar L, Spada A, Giannattasio G, (1987) Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas Nature 330(6148): 566-568.
- Ma ZY, Song ZJ, Chen JH, Wang YF, Li SQ, et al. (2015) Recurrent gain-offunction USP8 mutations in Cushing’s disease. Cell Res 25(3): 306-317.
- Ong OC, Hu K, Rong H, Lee RH, Fung BK (1997) Gene structure and chromosome localization of the G gamma c subunit of human cone G-protein (GNGT2). Genomics 44(1): 101-109.
- Tang H, Wei P, Duell EJ, Risch HA, Olson SH, et al. (2014) Genesenvironment interactions in obesity- and diabetes-associated pancreatic cancer: a GWAS data analysis. Cancer Epidemiol Biomarkers Prev 23(1): 98-106.
- Steenaard RV, Ligthart S, Stolk L, Peters MJ, van Meurs JB, et al. (2015) Tobacco smoking is associated with methylation of genes related to coronary artery disease. Clin Epigenetics 7: 54.
- Kim YH, Lee HC, Kim SY, Yeom YI, Ryu KJ, et al. (2011) Epigenomic analysis of aberrantly methylated genes in colorectal cancer identifies genes commonly affected by epigenetic alterations. Ann Surg Oncol 18(8): 2338-2347.
- Melmed S (2011) Pathogenesis of pituitary tumors. Nat Rev Endocrinol 7(5): 257-266.
- Vandeva S, Jaffrain Rea ML, Daly AF, Tichomirowa M, Zacharieva S, et al. (2010) The genetics of pituitary adenomas. Best Pract Res Clin Endocrinol Metab 24(3): 461-476.
- Dworakowska D, Grossman AB (2009) The pathophysiology of pituitary adenomas. Best Pract Res Clin Endocrinol Metab 23(5): 525-541.
- Simpson DJ, Hibberts NA, McNicol AM, Clayton RN, Farrell WE (2000) Loss of pRb expression in pituitary adenomas is associated with methylation of the RB1 CpG island. Cancer Res 60(5): 1211-1216.
- E Ezzat S, Zhu X, Loeper S, Fischer S, Asa SL (2006) Tumor-derived Ikaros 6 acetylates the Bcl-XL promoter to up-regulate a survival signal in pituitary cells Mol Endocrinol 20(11): 2976-2986.
- Zhu X, Asa SL, Ezzat S (2007) Ikaros is regulated through multiple histone modifications and deoxyribonucleic acid methylation in the pituitary. Mol Endocrinol 21(5): 1205-1215.
- Djamgoz MB Coombes RC Schwab A (2014) Ion transport and cancer: from initiation to metastasis. Philos Trans R Soc Lond B Biol Sci 369(1638): 20130092.
- Canny BJ, Rawlings SR, Leong DA (1992) Pituitary adenylate cyclaseactivating polypeptide specifically increases cytosolic calcium ion concentration in rat gonadotropes and somatotropes. Endocrinology 130(1): 211-215.
- Sato T, Nakamura Y, Shiimura Y, Ohgusu H, Kangawa K, et al. (2012) Structure, regulation and function of ghrelin. J Biochem 151(2): 119- 128.
