 Mini Review
Mini ReviewABSTRACT
Functional genomics is the study of gene function and interaction with other proteins that contribute to different metabolic pathways. The most commonly used approach for identifying gene function is to reduce or completely disrupt its normal expression pattern. Gene knock-down and gene knock-out are two reverse genetic approaches that are widely used for discovering gene function. RNAi-mediated gene silencing has been used as a magic bullet over a decade to disrupt gene expression in many organisms. However, new genome editing tools—especially CRISPR-based technologies—has become major breakthrough against RNAi dominance in reverse genetics studies. RNAi results in temporary silencing of gene expression (knock-down) but CRISPR/Cas system results in permanent gene silencing (knock-out). Both of this loss of function technology has their own advantages and limitations. The goal of a biological experiment is the crucial determining factor for choosing the right tool. The whole article compares the mechanism, advantage and disadvantages of these two technologies.
Abbreviations: ZFN: Zinc Finger Nuclease; TALEN: Transcription Activator-Like Effector Nucleases; CRISPR: Clustered, Regularly Interspaced Palindromic Repeat; RNAi: RNA Interference; CRISPR: Clustered Regularly Interspaced Short Palindromic Repeats; dsRNAs: Double-Stranded RNAs; siRNAs: Small Interfering RNAs; miRNAs: MicroRNAs; siRNAs: Short Interfering RNAs; TYMV: Turnip Yellow Mosaic Virus; RBSDV: Rice Black Streaked Dwarf Virus; HDR: Homology Dependent Repair; NHEJ: Non Homologous End Joining; ORF: Open Reading Frame
Introduction
The emergence of multiple genome editing technologies in recent years has revolutionized molecular biology research and enhanced the promises of modification of gene expression according to need of experiment. Functional genomics determine the relationship between genotype and phenotype on a genomic scale. The forward genetic approaches through random mutagenesis or viral transposons are difficult to perform on a large scale for modifying gene function. Development of RNAi and genome editing tools like ZFN (Zinc finger nuclease), TALEN (Transcription Activator-Like Effector Nucleases) and CRISPR (Clustered, Regularly Interspaced Palindromic Repeat) has overcome the technical barriers and has become popular reverse genetic tools for gene expression modification.
Fire and Mello discovered that injecting double-stranded RNA into C. elegans silenced gene sequence and produce phenotypes that revealed gene function Fire, et al. [1]. The repression of gene expression by double-stranded RNA is known as RNAi mechanism. Over the past decade, RNA interference (RNAi) has dominated various research experiments of transient gene expression repression at the post-transcriptional level through double stranded RNA Unniyampurath, et al. [2]. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and the CRISPRassociated protein 9 (Cas9) (CRISPR/Cas9) system of prokaryotes has been used for stable genetic modifications Boettcher, et al. [3]. CRISPR/Cas9 system has the ability to introduce heritable site-specific insertions and deletions in the eukaryotic genome Unniyampurath, et al. [2]. The increasing popularity and superior capabilities of CRISPR/Cas9 system can make it able to occupy the position of RNAi in field of molecular biology research. Each of these two loss-of-function technology has their own advantages and limitations, so choosing the right tool for the job mainly depend on the requirement of specific experimental design. Both RNAi and CRISPR-Cas9 have significant milestones in field of molecular biology. In this review along with determining superiority, the history, mechanism and application of these two technologies are described (Figure 1).
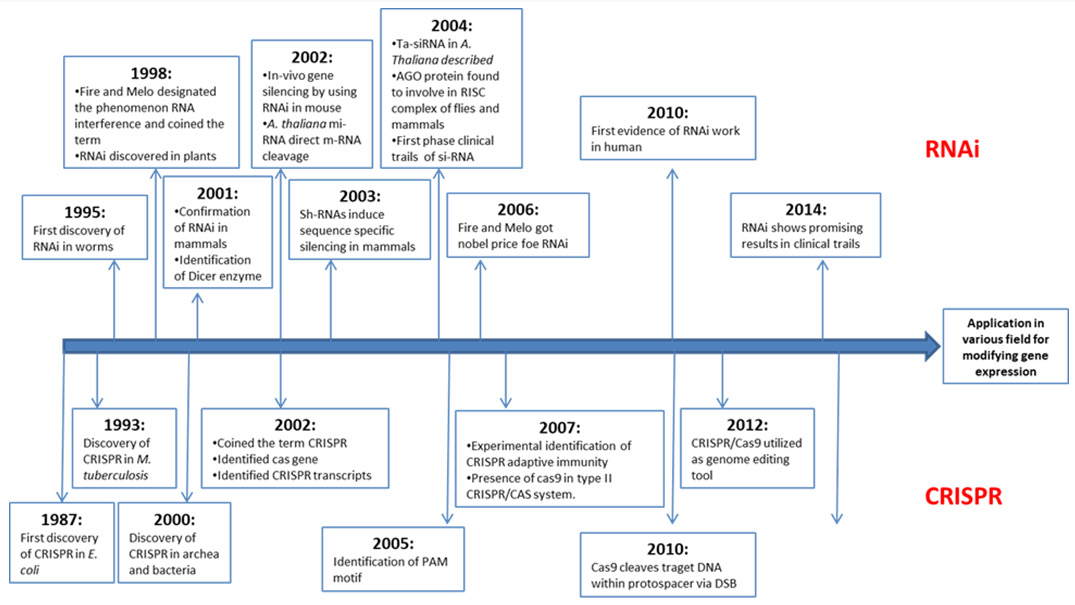
Figure 1: The timeline of milestones in RNAi and CRISPR.
RNA interference (RNAi)
In C. elegans, fire and his colleagues (1998) first coined the term RNAi. It is a new and strong method for halting mRNA gene expression. In mammalian cells, the Dicer which is a family of RNase III endonuclease cleaves longer dsRNA precursors to produce siRNAs. Dicer is a large endoribonuclease that processes double-stranded RNAs (dsRNAs), produces approximately 20 bplong small interfering RNAs (siRNAs) that act as effectors during RNA interference (RNAi) and excises microRNAs (miRNAs) from the precursors molecules that is assemble into RISC complex and direct this complex to complementary mRNA targets. Once binding with small RNA, RISC complex silence the targeted genes.
Components of RNAi
Dicer
Dicer enzymes are often less diversified in animals Meister, et al. [4] The RNase III enzyme family includes dicer enzymes, which recognise the ends of lengthy dsRNAs and cleave the RNA approximately 21 nt from the end Treiber, et al. [5]. Dicer-like enzymes have two catalytic RNase III domains that cleave both strands and leave two nucleotides 3′ overhangs due to their location on the dsRNA.
Arganoute
AGO proteins are highly specialised sRNA-binding modules that are important components of RISCs in silencing pathways. Small RNAs created during the initiation step are loaded into AGO proteins to direct sequence-specific regulation of gene expression, and AGO proteins perform the effector phase of silence.
RNA-Dependent RNA Polymerase (RDR) Proteins
Third component of RNAi pathway is RDR (RNA dependent RNA polymerase) which convert ssRNAs to dsRNAs, which are then processed by DCLs resulting in a new cycle of RNA silencing. RDRs are required for the synthesis of phased and repeat-associated siRNAs in sRNAs, but miRNAs and hairpin-derived sRNAs are RDRs independent RNAs. RDR activity was originally investigated in Chinese cabbage in 1971 and tomato cDNA was obtained in 1972. RDRs are the first components of plant small RNA biogenesis pathways to be identified and they are distinguished by a unique RNA-dependent RNA polymerase catalytic domain (RdRp).
Mechanism of RNA interference
RNA interference (RNAi) is a type of gene silence caused by the processing of double-stranded RNA (dsRNA) into short interfering RNAs (siRNAs). Both post-transcriptional and transcriptional gene silencing can be induced by RNAi. An enzyme resembling RNase III called Dicer which produces a 19–21 nucleotide duplex Hutvagner [6] siRNA with two nucleotide projections at its 3ʹ end via the cleavage of long dsRNAs. In RNAi pathway, the siRNA guide strand directs RISC to perfectly complementary RNA targets, which are then degraded. RNA degradation is induced by the PIWI domain of the Ago protein. siRISC identifies a completely complementary mRNA, resulting in Ago-catalyzed mRNA cleavage at a single duplex site. The functional siRISC is regenerated after cleavage, while the mRNA fragments are degraded further. Simultaneously transcribed RNAs (siRNAs) can recognise targets with poor complementarity.
RNAi as a Anti Viral Defence Mechanism
Plant antiviral biotechnology is now using RNA interference (RNAi). Artificial miRNA (amiRNA) is a robust biotechnology utilised in plants for gene silencing, and amiRNA engineering has been widely employed for the targeted down-regulation of endogenous genes in a variety of plants. The endogenous precursor of miRNA produced from the host has been widely exploited to substitute 21-nt long miRNA sequence with a region complementary to the target viral genome sequence. Multiple-target miRNAs can also effect multiple viruses at the same time. miRNA precursors which containing complementary sequences with Turnip yellow mosaic virus (TYMV) and the transgenic Arabidopsis expressing the recombinant miRNA precursors displayed specific resistance to these viruses Niu, et al. [7]. Based on the structure of the rice osa-MIR528 precursor. Sun, et al. [8] created three dimeric amiRNA precursor expression vectors that target the 3-proximal region of the CP genes of RSV and Rice black streaked dwarf virus (RBSDV). At a low temperature, the transgenic rice plants demonstrated great resistance to both RSV and RBSDV infection Sun, et al. [8].
RNAi as a Functional Genomics Approach and its Therapeutic Applications
In several animals, RNAi technology can be used to silence genes. In C. elegans, RNAi has been widely utilized for functional genomics. The majority of the 19,000 genes have been investigated at a high-throughput level. Ahringer and his colleagues created an RNAi library that contained 86 percent of C. elegans genes. This method has been tried and tested in a variety of additional model organisms. Mammalian cells have also been effectively treated using RNAi Hu, et al. [9]. In mammalian cells, many approaches for siRNA knockdown of specific genes have been used. In mammalian cells, DNA-vector-mediated RNAi silences genes selectively, while alternative expression methods are used for long-term silencing. For stable silence, the promoters of RNA polymerase (pol) II and III (U6 and H1, alone or together) have been employed.
CRISPR: (Clustered Regularly Interspaced Short Palindromic Repeats)
CRISPR is an adaptive immune mechanism of prokaryotic
organisms. They are a family of DNA sequences used to detect and
destroy the foreign invading DNA of bacterial infecting viruses
(bacteriophage) with the help of CRISPR asoociated nucleases
thereby preventing infection. Consequently, utilizing this innate
immune mechanism, so called the CRISPR technology, allows the
generation of desired modifications on specific target sequences in a
genome. This modification is utilized to introduce mutations barring
the need for traditional breeding programmes. The mutations are
induced by exploiting the DNA repair pathways of the cells either
through homology dependent repair (HDR) or non homologous
end joining (NHEJ) pathways. The mutations so induced changes
the Open reading frame (ORF) which in turn creates, gene knock
down and knock-offs thereby creating variation. The advanced
utilization of CRISPR is the ability to alter one specific nucleotide
into another which is known as base editing.
Types of CRISPR and associated nucleases used in CRISPR
cascade:
1. Type I: The Type I systems are encoded by a single operon
and consists of a signature cas3 gene which codes for a large
protein with a helicase activity. They possess a single stranded
DNA stimulated ATPase activity associated with the unwinding
of both DNA-DNA duplex and RNA-DNA duplex Sinkunas, et al.
[10].
2. Type II: The signature gene for this type of CRISPR system is
cas9 which encodes for a multi- domain protein combining all
the functions of effector complexes and cleavage of target DNA
Jinek, et al. [11]. This system also has the ability utilize the
cellular RNAse III and tracrRNA for processing the pre-crRNA
Deltcheva, et al. [12]. Usually this system contains two domains
in its nuclease namely the Ruv-C and the HNH domains which
are both required to cleave the DNA in its targeted site.
3. Type III: The signature gene associated with this system is
cas10 which encodes for a multi- domain protein. The protein
consists of a palm domain which is similar to polymerases and cyclases of the polB family Anantharaman, et al. [13]. This
type of CRISPR systems do not encode their own cas1 and cas2
genes but rather make use of crRNA produced from CRISPR
arrays associated with type I or type II systems Nickel, et al.
[14].
4. Type IV: This type of CRISPR system is generally fond in the
plasmids of bacterial genomes. They lack cas1 and cas2 genes
and also are not involved with any of the CRISPR arrays. This
has been identified as the only CRISPR/cas system with no
detectable CRISPR cassettes in the genome Makarova [15].
5. Type V: This type of CRISPR system comprises of rRNA and
cas12a. The Cas12a protein contains a Ruv-C endo-nuclease
domain which cleaves the non-target strand and the targeting
strand to form a DSB Gao, et al. [16].
Advantages of CRISPR
To start with, this technology has been deemed to be more accurate and target specific for editing the sequences within the genome, thereby becoming one of the most widely used tool to create variations and mutations Zhang, et al. [17]. It is also efficient and fairly easy to use as compared to other editing tools like TALENs (Transcription activator-like effector nucleases) and ZFNs (Zinc finger nucleases) which are expensive and difficult to handle due to its simplicity in CRISPR/cas programming Qiu, et al. [18]. The efficiency of inducing mutation is highest through CRISPR. It is possible to target more than one target site from a single CRISPR cassette also known as multiplex CRISPR Sakuma, et al. [19].
Challenges in CRISPR Technology
Even though there has been advancement in scientific technologies, bioethical values play a huge role in the technology linked with genome editing technologies Brokowski [20]. Even though the efficiency of creating mutation in targeted sites are highest in CRISPR as compared to other gene editing tools, the efficiency of developing mutations through HDR is fairly low in comparision to the NHEJ pathway. Using CRISPR technology often triggers cell apoptosis on the subjected tissue intended for editing the genes, due to DSB’s Hu, et al. [21]. During the designing of gRNA, optimum care should be taken so that there is no off- target effects when subjected to editing the target tissue.
RNAi vs. CRISPR
RNAi is the post-transcriptional gene silencing mechanism generally found in eukaryotes. Whereas CRISPR is the new age genome editing tool that naturally serves as defence barrier found in prokaryotes. Because of its simplicity and wider capability, the type II CRISPR/Cas9 system has been widely used as a robust and versatile tool for gene editing now a day. RNAi, the major dominating genetic tool ruling the molecular biology laboratory over a decade, has some advantages and disadvantages over CRISPR technology. The CRISPR/Cas system has been used to induce gene modification by knockout via DSBs and NHEJ repair, transcription repression (CRISPRi/ CRISPR Interference) and transcription activation (CRISPRa/ CRISPR activation) Boettcher, et al. [3]. RNAi is the mechanism of post-transcriptional repression of gene expression by ds-RNA. CRISPR/Cas system and RNAi are occupying several overlapping domains of molecular biology research, raising the possibility that in near future one may selectively dominate the other. The fundamental difference between RNAi and CRISPRCas9 is that RNAi causes knockdown of gene expression at the post-transcriptional level by targeting mRNA, whereas CRISPRCas9 causes knockout of gene expression by targeting DNA. The main determinable factor for choosing the right tool is the aim of experiment.
In some cases like slow protein degradation and absence of rate limiting factor transcript levels—as caused by knockdown approaches may not produce a loss-of-function phenotype Boettcher, et al. [3]. In that case complete knock out of gene expression is the need of the experiment. When temporary reduction of gene expression is the primary need of experiment, RNAi can be advantageous over CRISPR as stable modification of the genetic code is undesirable and si-RNA may be lost after some generation Moreira, et al. [22]. Sometimes the complete elimination gene function is detrimental to the cell rather than partial loss Boettcher, et al. [3]. CRISPR/Cas system makes it possible to eliminate all variants of transcripts of a gene by using sg-RNA Wang, et al [23]. The CRISPR/Cas9 technique is a precise genome editing which leads to sequence-specific desirable modifications. The ontarget efficiencies of sgRNAs and shRNAs are found to be similar in a large-scale screen of CRISPR and RNAi (4). Off-target effects are less in case of CRISPR than RNAi (4). Due to the small length of si-RNA (24 nt), it can silence non-target mRNAs with only limited sequence complementarity (7). RNAi can only cause post-transcriptional or transcriptional gene silencing, while variations of CRISPR can be involved in gene knockout, knock down, transcrioptional activation or repression Boettcher, et al. [3]. Unlike CRISPR, RNAi targets RNA transcripts in the cytoplasm not genomic DNA in the nucleus. RNAi can be used in species for which only transcriptome data exists [24- 29].
Conclusion
CRISPR/Cas9 and RNAi both are powerful and useful tools for gene manipulations. CRISPR/Cas9 is more flexible than RNAi due to its versatile application in induction of InDels, repression or activatation of gene expression, and causing heritable and nonheritable genomic changes Unniyampurath, et al. [2]. The discovery of Cpf1 enzyme has revolutionized the CRISPR mediated genome engineering. RNAi is having a unique space in diverse genetic applications. But the increasing advantages of CRISPR/Cas9 may dominate over RNAi in near future. CRISPRi is the biggest threat to RNAi. However, wider application of CRISPRi system is yet to be established at a genome scale. In near future predictabily, CRISPR/ Cas9 will rule molecular biology lab for modifying gene expression, while RNAi will likely to be cornered with restricted domains of applications.
References
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, et al. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391(6669): 806-811.
- Unniyampurath U, Pilankatta R, Krishnan MN (2016) RNA Interference in the Age of CRISPR: Will CRISPR Interfere with RNAi?. International journal of molecular sciences 17(3): 291.
- Boettcher M, McManus MT (2015) Choosing the right tool for the job: RNAi, TALEN, or CRISPR. Mol Cell 58(4): 575-585.
- Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, et al. (2004) Human Argonaute2 Mediates RNA Cleavage Targeted by miRNAs and siRNAs. Mol Cell 15(2): 185-197.
- Treiber T, Treiber N, Meister G (2019) Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol 20(1): 5-20.
- Hutvágner G, Zamore P (2002) A microRNA in a multiple-turnover RNAi enzyme complex. Science 297(5589): 2056-2060.
- Niu Q W, Lin SS, Reyes JL, Chen KC, Wu HW, et al. (2006) Expression of artificial microRNAs in transgenic Arabidopsis thaliana confers virus resistance. Nat Biotechnol 24(11): 1420-1428.
- Sun L, Lin C, Du J, Song Y, Jiang M, et al. (2016) Dimeric artificial microRNAs mediate high resistance to RSV and RBSDV in transgenic rice plants. Plant Cell Tiss Organ Cult 126(1): 127-139.
- Hu G, Kim J, Xu Q, Leng Y, Orkin SH, et al. (2009) A genome wide RNAi screen identifies a new transcriptional module required for self renewal. Genes Dev 23(7): 837-848.
- Sinkunas T, Gasiunas G, Fremaux C, Barrangou R, Horvath P, et al. (2011) Cas3 is a single‐stranded DNA nuclease and ATP‐dependent helicase in the CRISPR/Cas immune system. The EMBO journal 30(7): 1335-1342.
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, et al. (2012) A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, et al. (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471(7340): 602-607.
- Anantharaman V, Iyer LM, Aravind L (2010) Presence of a classical RRM-fold palm domain in Thg1-type 3'-5'nucleic acid polymerases and the origin of the GGDEF and CRISPR polymerase domains. Biology direct 5: 43.
- Nickel L, Weidenbach K, Jäger D, Backofen R, Lange SJ, et al. (2013) Two CRISPR-Cas systems in Methanosarcina mazei strain Gö1 display common processing features despite belonging to different types I and III. RNA biology 10(5): 779-791.
- Makarova KS, Koonin EV (2015) Annotation and classification of CRISPR-Cas systems. CRISPR, p. 47-75.
- Gao P, Yang H, Rajashankar KR, Huang Z, Patel DJ (2016) Type V CRISPR-Cas Cpf1 endonuclease employs a unique mechanism for crRNA-mediated target DNA recognition. Cell research 26(8): 901-913.
- Zhang JH, Adikaram P, Pandey M, Genis A, Simonds WF (2016) Optimization of genome editing through CRISPR-Cas9 engineering. Bioengineered 7(3): 166-174.
- Qiu XY, Zhu LY, Zhu CS, Ma JX, Hou T, et al. (2018) Highly effective and low-cost microRNA detection with CRISPR-Cas9. ACS synthetic biology 7(3): 807-813.
- Sakuma T, Nishikawa A, Kume S, Chayama K, Yamamoto T (2014) Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Scientific reports 4(1): 5400.
- Brokowski C (2018) Do CRISPR germline ethics statements cut it?. The CRISPR journal 1(2): 115-125.
- Hu Z, Yu L, Zhu D, Ding W, Wang X, et al. (2014) Disruption of HPV16-E7 by CRISPR/Cas system induces apoptosis and growth inhibition in HPV16 positive human cervical cancer cells. BioMed research international 2014: 612823.
- Moreira D, Pereira AM, Lopes AL, Coimbra S (2020) The best CRISPR/Cas9 versus RNA interference approaches for Arabinogalactan proteins’ study. Mol Biol Rep 47(e3647): 3.
- Wang HY, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, et al. (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153(4): 910-918.
- Astier-Manifacier S, Cornuet P (1971) RNA-dependent RNA polymerase in Chinese cabbage. Biochim Biophys Acta Nucleic Acids Protein Synth 232(3): 484-493.
- Carthew RW, Sontheimer EJ (2009) Origins and Mechanisms of miRNAs and siRNAs. Cell 136(4): 642-655.
- Fraser AG, Kamath RS, Zipperlen P, Martinez-compos M, Sohrmann M, et al. (2000) Functionla genomic analysis of C.elegans chromosome 1 by systematic RNA interference. Nature 408(6810): 325-330.
- Vaucheret H (2008) Plant Argonautes. Trends Plant Sci 13(7): 350-358.
- Smith I, Greenside PG, Natoli T, Lahr DL, Wadden D, et al. (2017) Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map. PLOS Biol 15(11): e2003213.
- Sigoillot FD, King RW (2011) Vigilance and Validation: Keys to Success in RNAi Screening. ACS Chem Biol 6(1): 47-60.
