 Case Report
Case Report







Abstract
Mitochondrial 2-methylacetoacetyl-CoA thiolase deficiency (OMIM#203750, *607809) is a rare inherited disorder of isoleucine amino acid catabolism and ketone body metabolism. Clinical manifestations of this disorder are nonspecific and similar to the other organic acidurias. 2-methylacetoacetyl CoA thiolase encodes by ACAT1 gene and acts at the end stage of isoleucine amino acid catabolism pathway. Herein we report a 7yr-old boy with beta-ketothiolase deficiency as a cause of a novel compound heterozygote mutations [c.293G>A; (p. Gly98Asp) and c. (120+1_121-1) _ (334+1_335-1) del] in ACAT1 gene. Early diagnosis and treatment of this treatable neurometabolic disorder can prevent from neurologic sequels during ketoacidosis episodes.
Introduction
2-methylacetoacetyl CoA thiolase enzyme acts at the end stage of isoleucine amino acid catabolism pathway and is involved in the ketone body metabolism [1] (Figure 1). 2-methylacetoacetyl CoA thiolase deficiency (which is also known as Beta-ketothiolase deficiency, T2 deficiency, 3-oxothiolase deficiency, alpha-methyl acetoacetic aciduria, 2-methyl-3-hydroxybutyric acidemia, and mitochondrial acetoacetyl-CoA thiolase deficiency) is an autosomal recessive inborn error of metabolism and classified as a ketolysis disorder or organic aciduria disorder. Beta-ketothiolase caused by defects in the acetyl-CoA acetyltransferase 1 (ACAT1) gene. This gene is located on 11q22.3-23.1 and contains 12 exons that span approximately 27kb [2]. Clinical manifestations of this disorder usually appear between the first 6 to 24 months of life and include intermittent ketoacidosis attacks, vomiting, dyspnea and neurological symptoms such as lethargy, loss of consciousness, hypotonia, chorea and/or athetosis movement. Fruity or acetone odor on breathing indicates ketoacidosis. These episodes almost occur after severe stress such as infections, increased protein intake and prolonged fasting [3].
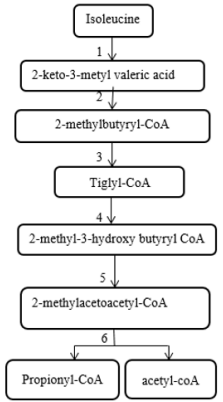
Figure 1: The pathway of isoleucine metabolism
1. Branched chain aminoacid aminotransferase
2. Branched chain alpha ketoacid dehydrogenase
3. short/branched chain acyl-CoA dehydrogenase deficiency
4. hydratase
5. 2-methyl-3-hydroxybutyryl- CoA dehydrogenase
6. β-ketothiolase
Chronic neurological complications such as developmental delay and movement disorders due to basal ganglia involvement could be seen independently to ketoacidosis attacks [4]. Patients are symptom-free between metabolic crises and the frequency of ketoacidosis attacks in them decreases with age [3]. The diagnosis of this disorder is straightforward and includes an elevated level of 2-methyl-3-hydroxybutyric acid, 2-methylacetoacetic acid and tiglylglycine in urine organic acids analysis and elevated levels of acylcarnitines C5:1 and C5OH in blood acyl carnitine profile analysis [5]. Confirmatory test is molecular analysis. Symptomatic treatment of beta-ketothiolase deficiency during attacks of metabolic decompensation is supportive and includes fluid therapy and correction of metabolic acidosis (by parental bicarbonate) and correction of electrolyte disturbances. Oral or parenteral carnitine is also useful. For long-term treatment; avoiding of prolonged fasting (frequent feeding, IV glucose during vomiting or severe illness), limited intake of protein to about 1.5-2gr/kg/day, avoiding of highfat diet (similar to ketogenic diet) and L-carnitine supplementation have been recommended [6]. Here, we report a 7yr-old boy with frequent metabolic decompensating episodes presented during infancy suspicious to beta-ketothiolase deficiency. Compound heterozygote mutations in ACAT1 gene were identified in this patient by with whole exome sequencing (WES).
Clinical Presentation
A 6 months old boy was admitted to Children Medical Center Hospital, Tehran, Iran due to decreased level of consciousness, hypotonia and choreoathetoid movements of hands following an upper respiratory tract infection. He was the first child of a consanguineous marriage and was born through normal vaginal delivery with birth weight, height, and head circumference of 3350 grams, 48cm and 36cm, respectively. Initial biochemical and metabolic studies showed just a compensated high anion gap metabolic acidosis (PH=7.06 and HCO3- =8.5μmol/L in venous blood gas), a mild lactic acidosis and 2+ positive urine ketone. His parents reported an abnormal movement in both hands of their child started at the age of 5.5 months which followed by the first ketoacidosis attack at the age of 6 months. He was discharged from the hospital but after this acute metabolic crisis, the patient developed axial hypotonia though that he was not able to hold his neck anymore. At the age of 12 months, patient was admitted at hospital again due to another similar episode of disease with decreased level of consciousness in addition of upper limbs dystonia and choreiform movements following age-appropriate vaccination program.
Laboratory tests showed severe metabolic acidosis and increased plasma lactate level (35mg/dl; normal range: 2-20mg/dl) but serum ammonia level, serum amino acids chromatography (by HPLC method) and acylcarnitine profile [measured by tandem mass spectrometry (MS/MS)] were normal. Urine organic acid analysis [by GC/MS method (Gas chromatography-mass spectrometry)] indicated an elevated level of 3-hydroxy-2-methyl butyric acid, 3-keto-2-methyl butyric acid and tiglylglycine. He was discharged from hospital and a metabolic cocktail (Biotin, L-carnitine, coenzyme Q10, and vitamin B1, vitamin B12) was prescribed for him. In addition, he was referred to neurology clinic for evaluation of motor delay and limbs dystonia. On physical examination at the age of 13 months (age of referral to Myelin Disorders Clinic), his weight and head circumference were 9500gr (10th percentile) and 46cm (25th percentile), respectively. Neurological examinations at this age showed motor delay, mild to moderate axial hypotonia, normal knee- and ankle-deep tendon reflexes and choric movements of both hands. The patient was not able to sit independently, but neck holding had been improved significantly based on available medical records.
Cognition and eye pursuit movements were normal. There was no history of seizure. A neurometabolic disorder was suspected due to presence of clinical symptoms of cerebral hypotonia, upper limbs dystonia and choreoathetiod movements and history of two metabolic decompensation episodes with high anion gap metabolic acidosis. The first brain magnetic resonance imaging (MRI) at the age of 13 months was normal. The pattern of urine organic acid profile remained abnormal suspected to Beta-ketothiolase deficiency in follow up visits even when the patient was not clinically in metabolic decompensation state. In follow-up visits, complete pattern of independent walking was achieved at the age of 4 years and the patient was able to make three-word sentences at this age. Review of medical records until age of 5 years revealed additional three times short course hospitalization of our patient due to illness, infrequent vomiting and mild metabolic acidosis following acute viral gastroenteritis or upper viral respiratory infections during summer or winter months around the year. After the last admission at the age of 6 years, the Shohl’s solution was added to the metabolic cocktail in drug regimen of the patient. During the last two years until age 8years, the patient has not had any hospital admission or acute metabolic decompensation episode.
At the last visit at the age of 8 years we found out that the patient goes to school with normal speech and cognition but mild imbalance gait and choreic movements of the upper limbs. Weight, length and head circumference at this age were 25Kg (50th percentile), 125 cm (25th percentile) and 50 cm (50th percentile), respectively. Metabolic cocktail drugs and Shohl’s solution were discontinues gradually from the age of 7years except for IL-carnitine supplementation, low protein (2gr/kg/day) and a normal fat diet. According to the medical history and laboratory information, diagnosis of 2-methyl acetoacetyl CoA thiolase deficiency was considered for this patient, but the family was not able to perform genetic analysis until the age of 7 years old. WES was performed on the patient using Nextera® Rapid Capture enrichment kit followed by Next generation sequencing (NGS) on a Nextseq500, Illumina machine. Evaluation was focused on the coding exons along with flanking +/-15 intronic bases within the captured region. All the disease-causing variants reported in HGMD® or in ClinVar in addition to all variants with minor allele frequency (MAF) of less than 1% in gnomAD database were considered. Result showed compound heterozygote mutations, including a previously reported heterozygote deletion of exon 3 and 4, c. (120+1_121-1) (334+1_335-1) del, and a novel heterozygous mutation c.293G>A(p.Gly98Asp) in the other allele of the ACAT1 gene in the index case. Genetic analysis of the parents revealed that his mother was heterozygous for the deletion of exon 3 and 4 and his father was carrier for the c.293G>A mutation.
Discussion
Beta-ketothiolase deficiency or 2-methylacetoacetyl CoA thiolase deficiency is a rare hereditary and treatable neurometabolic disorder with various clinical manifestations. So far, about 135 cases with beta-ketothiolase deficiency have been reported in the literature with similar symptoms to our case such as intermittent ketoacidosis attacks in some patients and neurological symptoms (confusion, lethargy, decrease of consciousness level, movement disorder, and coma) in some others [6]. The most common age of first attack in the other reports similar to our patient are between 6 and 36 months because children are getting exposed to common infections for the first time during this period [7]. Some patients have been diagnosed before manifesting any symptoms by newborn screening [8]. Daum, reported the first case of Beta-ketothiolase deficiency in 1971, then after several different mutations of ACAT1 gene have been reported in the literature [1]. Based on Human Genome Mutation Database, around 100 mutations have been identified so far in ACAT1 gene and most of them are missense or nonsense mutations.
Some of them are common in a specific population; for instance, p.R208* and c.1006-11G>C in the Vietnamese and p.M193R in Indian population [7,9]. We found in our patient a novel heterozygote mutation c.293G>A in ACAT1 (NM. _000019.3) gene associated with an amino acid change of p. Gly98Asp affecting a moderately conserved amino acid in N-terminal thiolase-domain. This mutation is neither listed in mutation databases nor described in the literature and based on Mutation Taster, Poly-Phen2, Provean and LRT is classified as disease-causing, whereas SIFT and GVGD classified it as benign. Additionally, our patient carries a heterozygous deletion c. (120+1_121-1) _ (334+1_335-1) of exon 3 and 4 of ACAT1 gene which has been previously reported in a patient with 2-methylacetoacetyl CoA thiolase deficiency [2]. The non-equal homologous recombination between Alu sequences in ACAT1 gene has been proposed as the reason for occurring the deletion of exon 3 and 4. Alu repeats are frequently distributed throughout the human genome and unequal crossing over between Alu sequences are associated with positional deletion and insertion events. 32.7% of the whole beta-ketothiolase gene is comprised of Alu sequences.
Intron 2 has three and intron 4 has two Alu sequences. Probably a non-equal homologous recombination between Alu repeats in intron 2 and 4 leads to deletion of exons 3 and 4. This exon skipping leads to generation of a truncated mRNA which is subject to nonsense-mediated mRNA decay [2]. Deletion of exon 3 and 4 was heterozygously found in his mother, whereas his father was a carrier of the c.293G>A; Thereby confirming the compound heterozygote pattern and the diagnosis of 2-methylacetoacetyl-CoA thiolase deficiency in our patient. Two other patients with beta-ketothiolase have been reported from Iran so far including a novel homozygote mutation c.664A>C (p. Ser222Arg) in ACAT1 gene [10-12]. As Beta-ketothiolase deficiency is considered a treatable neurometabolic disorder, early diagnosis in our case resulted in a fairly good prognosis and a near normal age appropriate life style [2].
Conclusion
In conclusion, although 2-methylacetoacetyl CoA thiolase deficiency is a rare neurometabolic disease, the pediatrician should consider this disorder in the setting of ketoacidosis attack with or without neurological symptoms such as unexplained intellectual disability or movement disorders. Adequate treatment may prevent neurological and other serious complications over the time.
Acknowledgment
We thank our patient’s family for participating in this study.
References
- Duam RS, Lamm PH, Mamer OA, Scriver CR (1971) A new disorder of isoleucine catabolism. Lancet 2 (7737): 1289-1290.
- Fukao T, Aoyama Y, Murase K, Hori T, Harijan RK, et al. (2013) Development of MLPA for human ACAT1 gene and identification of heterozygous Alu-mediated deletion of exon 3 and 4 in a patient with mitochondrial acetoacetyl-CoA thiolase (T2) deficiency. Molecular genetics and metabolism 110(1): 184-187.
- Fukao T, Scriver CR, Kondo N (2001) The clinical phenotype and outcome of mitochondrial acetoacetyl-CoA thiolase deficiency (β- ketothiolase or T2 deficiency) in 26 enzymatically proved and mutation-defined patients. Mol Genet Metab 72(2): 109-114.
- Paquay S, Bourillon A, Pichard S, Benoist JF, de Lonlay P, et al. (2017) Mitochondrial acetoacetyl-CoA thiolase deficiency: basal ganglia impairment may occur independently of ketoacidosis. J Inherit Metab Dis 40(3): 415-422.
- Duam RS, Scriver CR, Mamer OA, Edgard Delvin, Peter Lamm, et al. (1973) An inherited disorder of isoleucine catabolism causing accumulation of α-methylacetoacetate and α-methyl-β–hydroxybutirate and intermittent metabolic acidosis. Pediatr Res 7: 149-160.
- Toshiyuki Fukao, Hideo Sasai, Yuka Aoyama, Hiroki Otsuka, Yasuhiko Ago, et al. (2018) Recent advances in understanding beta-ketothilase (mitochondrial acetoacetyl –CoA thiolase T2) deficiency. Journal of Human Genetics 64(2): 99-111.
- Nguyen KN, Abdelkreem E, Colombo R, Hasegawa Y, Can NTB, et al. (2017) Characterization and outcome of 41 patients with betaketothiolase deficiency: 10 years’ experience of a medical center in northern Vietnam. J Inherit Metab Dis 40(3): 395-401.
- Catanzano F, Ombrone D, Stefano C, Rossi A, Nosari N, et al. (2010) The first case of mitochondrial acetoacetyl-CoA thiolase deficiency by expanded newborn metabolic screening in Italy: the importance of an integrated diagnostic approach. J Inherit Metab Dis 33(3): S91-S94.
- Abdelkreem E, Akella RRD, Dave U, Sane S, Otsuka H, et al. (2017) Clinical and mutational characterizations of ten Indian patients with beta-ketothiolase deficiency. JIMD Rep 35: 59-65.
- Rahim Vakili, Somayyeh Hashemian (2018) A Novel Mutation of Betaketothiolase Deficiency: The First Report from Iran and Review of Literature. Iran J Child Neurol 2018 summer 12(3): 113-121.
- Kobra Shiasi Arani, Babak Soltani (2014) First Report of 3-Oxothiolase Deficiency in Iran. Int J Endocrinol Metab 12(2): e10960.
- Van Karnebeek CD, Stockler S (2012) Treatable inborn errors of metabolism causing intellectual disability: A systematic literature review. Mol Genet Metab 105(3): 368-381.
