info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: January 08, 2018; Published: January 16, 2018
*Corresponding author: Shaziya Yasmeen Sayeed, Bhupal Nobles Institute of Pharmaceutical Sciences, Udaipur (Raj.), India
DOI: 10.26717/BJSTR.2018.02.000662
Bioburden is measure of microbial contamination or microbial load; the amount of microorganisms contaminating an object. Each lot of a component, drug product container, or closure with potential for microbiological contamination that is frightful in perspective of its planned utilize might be subjected to microbiological tests before utilize. Appropriate written procedures, designed to prevent objectionable microorganisms in drug products not required to be sterile, shall be established and followed. Microbiological monitoring of the manufacturing environment can serve as an adjunct to control and generally is a qualitative assessment tool in a properly implemented formal risk-based microbiological control program. A monitoring program corresponding with the product bioburden risk can help confirm the effectiveness of microbiological controls and may facilitate early detection of potential problems. The techniques utilized for microbial monitoring should be capable of isolating the numbers and types of organisms that have been deemed significant relative to in-process system control and product impact for each individual system.
Keywords: Bioburden; Microbiological Testing; Sterility; Sterile Products; Pharmaceuticals; Quality of Drugs
Abbreviations: WFI: Water for Injection; EM: Environmental Monitoring; DP: Drug Product
The microbiological quality of drugs is necessary for their efficacy and the safety of the patient s, because microbial contamination of drugs causes immediate adverse effects on patient health in terms of morbidity and mortality as well as longterm adverse effects, such as cancer, autoimmune diseases, and other diseases. [1-3]. Additionally, microbes can also alter the chemistry and pharmacology of drugs, with a potential adverse impact on their effectiveness due to the breakdown of the active ingredients as well as on their safety due to the toxicity of potential degrading products. Therefore, control of microbes in drugs is essential, either by assuring absence of microbes in sterile drugs that are administered parenterally and applied to sensitive tissues or by controlling microbial bioburden to appropriate levels for nonsterile drugs that are administered to regions rich in microbial flora with physical or immunological barriers to infections.
For sterile drugs, microbes are essentially eliminated by terminal sterilization which include heat or irradiation of final containers or by employing an aseptic manufacturing process where terminal sterilization is not possible, specifically for most biologics. Assurance of the absence of bacterial, yeast, and fungal contaminants is provided by the sterility test for sterile drugs [4]. For non-sterile drugs, bioburden due to aerobic bacteria, yeast, and fungi and absence from objectionable microorganisms, as required, is controlled to appropriate levels based on product attributes, route of administration; oral, intranasal, topical, anal, vaginal, etc and target patient population. Non-sterile drugs are tested for total aerobic bacteria, yeast, and fungi by the bioburden or microbial limit test and for the absence of objectionable organisms [5-7] (Table 1).
Table 1: Differences between sterile drugs and non-sterile drugs [8].
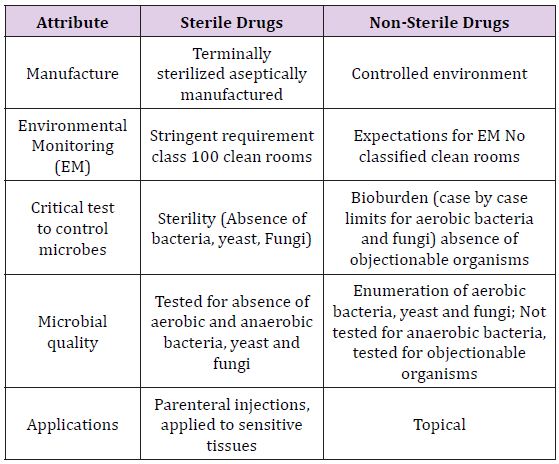
In the CMC audit process, manufacturing process controls are reviewed to assure consistent advances are set up to exhibit a condition of control that will assure the nature of the product. With trial of microbial burdens, bioburden control can be exhibited for a few purposes. Terminally sterilized drugs that utilize bioburdenbased cycles require process control testing to archive that the presterilization microbial load was sterilization the range set up for the sterilization procedure. Regardless of whether the sterilization is bioburden-based or utilizes an overkill approach, all items should incorporate controls that show the bulk and its ingredients are not adulterated.
First, although the drug may be rendered sterile by processing such as an overkill heat cycle, microbial by-products can present different materials that are regularly not recognized in routine testing. Second, microbial contamination can debase the product by changing pH or altering the active pharmaceutical ingredient. Third, microbial count data can be useful to indicate that process control is changing from its desired point. As a process control, bioburden testing is part of the general quality framework for sterile generation. Components of a parenteral drug solution should be tested as necessary to assure they are not adding an inordinate microbial load to the bulk. Also, since the most widely recognized component of a parenteral product is water for injection (WFI), it appears to be sensible that the other components would have a lesser impact on the bulk bioburden.
Since the recommended level of microorganisms in WFI is 10CFU/100mL [8,9], if the mass surpasses that farthest point, then at that point it is sensible to ask where the extra microorganisms are originating from. As on account of certain raw materials derived from non-synthetic sources, these may have unavoidably great number of organisms and other testing would be needed to assure the safety of the finished product.
Microbiological quality should be incorporated into the drugs by understanding sources of contamination, environmental conditions, and product attributes that support development of microorganisms. Microbiological quality for sterile drug is assured by utilizing a powerful environmental monitoring (EM) program, suitable microbiological testing at different stages or intermediate products during manufacture, including the final drug product (DP) and utilizing approved manufacturing processes (eg, aseptic manufacturing processes, container closure studies, media fill studies, etc). During routine manufacture of sterile drugs utilizing aseptic manufacturing forms, EM is a fundamental and basic part to exhibit the condition of control of the facility, giving data on the microbial nature quality of manufacturing and testing environments. This is an imperative component for sterility affirmation of sterile drugs. There are various direction archives and controls on the EM parts of produce of sterile drugs [10-12].
Microbiological quality of non sterile drugs is imperative, as well, and can be assured through determination of appropriate controls through a risk analysis process. Numerous sterile medications have certain components or intermediate products that are classified as non-sterile and are manufactured like non-sterile drugs. Therefore, understanding the risk of introduction of microorganisms and their products (such as toxins and proteases) during manufacture of non-sterile drugs, and proposed utilization of the drug in a target population (such as use of vaccines in healthy individuals) are vital considerations in choosing a manufacturing process-sterile or nonsterile. There are potential and a need to control and monitor the environment for manufacture of non-sterile drugs, intermediate products, or components [13-15] (Table 2).
Table 2: The Incubation Requirement for Microbial Tests Used in Drug Manufacturing [16].
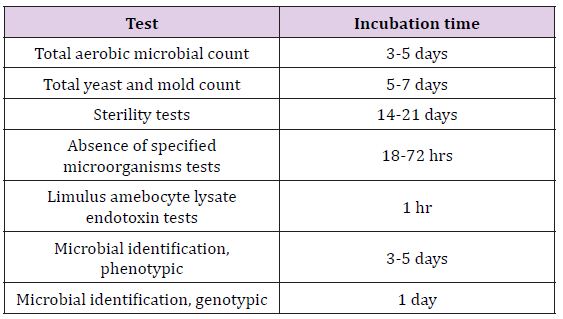
Sterility testing of media, in-process material, and final products must be performed during the manufacture of pharmaceuticals and medical devices and these are conducted by the following methods:
Sterility testing of unprocessed and final bulk, final vials, is normally performed by directly inoculating the test article into 2 distinct kinds of media that help the development and growth of aerobic and anaerobic bacteria, respectively. Test articles are incubated for 14 days followed by testing for microbial contaminants. Fungi are also detected.
Benefits:
a. Small volume required
b. Replicate samples can be tested
c. Preferred method for materials that cannot be easily filtered
To address issues for example large volumes of test article and the potential presence of inhibitors of microbial growth on samples, membrane filtration are used to test for enable sterility testing. In this strategy, bulk articles or effluent from vials of final product are passed through a membrane filter designed to retain microbial contaminants. The filters are rinsed to remove inhibitors and then incubated in two types of media and assayed exactly as in the direct inoculation method described above.
Benefits:
a. Enables testing of large sample volumes
b. Preferred method for products that can be filtered
c. Can remove inhibitory substances by rinsing the filter membrane with a suitable agent
Medical devices, solid dose forms, ointments, and creams may be directly immersed in growth media to test for bacteriostatic and fungistatic activity. Microbial detection is performed in a comparable way to direct inoculation after 14-day incubation. For test articles produced by non-aseptic manufacturing processes, a bioburden (or microbial limits) identification assay should be performed. Besides, it is frequently important to evaluate a nonsterile test article according to U.S. FDA and EP recommendations and guidelines for “objectionable organisms.” A microbial limit approach is recommended for the evaluation for objectionable organisms [16,17].
Microbiological monitoring of the manufacturing environment can serve as an adjunct to control and generally is a qualitative assessment tool in a properly implemented formal risk-based microbiological control program. A monitoring program corresponding with the product bioburden risk can help confirm the effectiveness of microbiological controls and may facilitate early detection of potential problems. The techniques utilized for microbial monitoring should be capable of isolating the numbers and types of organisms that have been deemed significant relative to in-process system control and product impact for each individual system.

























