info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map

Received: May 31, 2017; Published: June 15, 2017
Corresponding author: Leandro Bueno Bergantin, Laboratory of Autonomic and Cardiovascular Pharmacology, Department of Pharmacology, Escola Paulista de Medicina, Universidade Federal de São Paulo (UNIFESP), Phone-55 11 5576-4973, Rua Pedro de Toledo, 669 – Vila Clementino, São Paulo-SP, CEP: 04039-032, Brazil
DOI: 10.26717/BJSTR.2017.01.000131
Amyotrophic lateral sclerosis (ALS) is a devastating neurological disease characterised by progressive muscular paralysis reflecting selective degeneration and death of motor neurons in the primary motor cortex, brainstem and spinal cord. Several evidences suggest the involvement of the imbalance of the intracellular Ca2+ homeostasis in the death of motor neurons in ALS. It is now recognized that the interaction between intracellular signaling pathways mediated by Ca2+ and cAMP (Ca2+/cAMP signalling interaction) plays as a key role in several cellular processes of mammalians, including neurotransmission and neuroprotection.
Our previous studies have indicated that the pharmacological modulation of the Ca2+/cAMP signalling interaction by the combined use of the Ca2+ channel blockers (CCBs) and drugs that increment of the intracellular concentration of cAMP (cAMP-enhancer compounds) can increase neurotransmission, and stimulate neuroprotective response in neurodegenerative diseases. We have proposed that the pharmacological modulation of Ca2+/cAMP signalling interaction could open a new avenue for the drug development more effective and safer for treating neurodegenerative diseases, including ALS. In this review article, we discuss the perspectives of the pharmacological modulation of the Ca2+/ cAMP signalling interaction as a new therapeutic strategy for ALS.
Keywords: Ca2+/cAMP signaling interaction; Neurodegenerative diseases; Amyotrophic lateral sclerosis (ALS)
Described in 1874 by Jean-Martin Charcot, amyotrophic lateral sclerosis (ALS) is a devastating neurological disease characterized by progressive muscular paralysis reflecting selective degeneration of motor neurons in the primary motor cortex, brainstem and spinal cord Mathis et al. [1]. “Amyotrophy” refers to the atrophy of skeletal muscle fibers, which are denervated as their corresponding anterior horn cells degenerate, leading to muscle weakness; and “Lateral sclerosis” refers to hardening of the anterior and lateral corticospinal tracts as motor neurons in these areas degenerate, and are replaced by gliosis Rowland et al. [2]. ALS is also regarded as a multisystem degeneration with various other signs such as cognitive impairment (sometimes front temporal dementia), extra pyramidal features, postural abnormalities, and even small fiber neuropathy, and mild oculomotor disturbance Mathis et al. [1].
ALS is a neurodegenerative disorder that affects both upper (UMN) and lower (LMN) motor neurons. Muscle paralysis is progressive and leads to death due to respiratory failure within 2-5 years Mathis et al. [1]. The incidence of ALS is about 2 per 100,000 person/years, and its prevalence is about 5 per 100,000 persons Chiò et al. [3]. ALS can affect people at any age, but the peak age of onset is 55 to 70 years, with a male predominance (male: female ratio of about 3:2) Chiò et al. [3].
The causes of ALS are only partly known, but they include some environmental risk factors as well as several genes that have been identified as harbouring disease-associated variation Mathis et al. [1], Wang et al. [4]. Understanding what causes ALS, or influences survival, is crucial for the development of effective treatments. Significant advances have been made in understanding the genetic and environmental components of the disease. In accordance with the Amyotrophic Lateral Sclerosis Online Genetics Database (ALSoD), there are more than 25 genes in which an association with ALS has been replicated, with the rate of gene discovery doubling every 4 years. Most ALS cases are sporadic, but 5–10% of cases are familial, and of these 20% have a mutation of the SOD1 gene, and about 2–5% have mutations of the TARDBP (TDP-43) gene. Two percent of apparently sporadic patients have SOD1 mutations, and TARDBP mutations also occur in sporadic cases. There is mixed evidence for the involvement of chemicals, such as heavy metals, ambient aromatic hydrocarbons, pesticides, and cyanotoxins Sutedja et al. [5], Delzor et al. [6], Malek et al. [7], Bozzoni et al. [8], Rooney et al. [9].
Although the causes of ALS are not fully understood, some evidences suggest that the loss of motor function results from motor neurons death determined by imbalance of the intracellular Ca2+ homeostasis that causes cytosolic Ca2+ overload IIijic et al. [10], Wu & Wen [11]. The cytosolic Ca2+ overload is also involved in the dysregulation of glutamatergic signaling and alterations in neuronal toxicity in ALS Doble [12]. This glutamatergic excitotoxic hypothesis has given rise to an extremely active research field aimed at developing neuroprotective drugs blocking the excitotoxic process, such as riluzole Doble [12]. Riluzole is the only medication that has proved a modest effect in the survival of ALS patients. Oral administration of riluzole (100 mg daily) improves the 1-year survival by 15%, and prolongs survival by 3 months (after 18 months treatment), with a clear dose response Doble [12]. Longterm use of riluzole was associated with a better prognosis for ALS patients, whereas short-term use had little effect on survival Doble [12]. Then, other neuroprotective pharmacological strategies had been evaluated.
It is well recognized that the interaction between intracellular signalling pathways mediated by Ca2+ and cAMP (Ca2+/cAMP signalling interaction) plays as a key role in several cellular processes of mammalians, including neurotransmission and neuronal death Caricati-Neto et al. [13], Bergantin & Caricati-Neto [14]. Our previous studies have indicated that the pharmacological modulation of the Ca2+/cAMP signalling interaction by the combined use of the Ca2+ channel blockers (CCBs) and drugs that increment of the intracellular concentration of cAMP (cAMPenhancer compounds) can increase neurotransmission, and stimulate neuroprotective response in neurodegenerative diseases. Then, the pharmacological modulation of Ca2+/cAMP signalling interaction could open a new avenue for the drug development more effective and safer for treating neurodegenerative diseases, including ALS. In this review article, we discuss the perspectives of the pharmacological modulation of the Ca2+/cAMP signalling interaction as a new therapeutic strategy for ALS.
For understanding the cellular role of the Ca2+/cAMP signalling interaction, we should return to the past. Indeed, the concept of stimulus-secretion to explain neurotransmitters release has been achieved from ingenious experiments performed in the 1960s Douglas & Rubin [15]. From their concepts, it was showed in the 1970s that an increase in the cytosolic Ca2+ concentration ([Ca2+] c) is a fundamental requirement to start transmitter release Baker & Knight [16]. In addition, the unquestionable result showing a direct relationship between neurotransmitter release and elevation in [Ca2+]c came from the fundamental experiments made by the Nobel laureate Erwin Neher & Zucker [17]. Thus, by reducing extracellular Ca2+ through blocking Ca2+ channels, we should have a reducing in the neurotransmitter release. However, many reports have demonstrated that L-type CCBs, in concentrations below 1 μmol/L, could induce neurotransmitter release Kreye & Luth [18], French & Scott [19], Moritoki et al. [20]. In addition, many reports have demonstrated that the elevation of cytosolic cAMP concentration ([cAMP]c) enhances transmitter release at several synapses in autonomic nervous system of mammalians Chern et al. [21]. Recently, we demonstrated that Ca2+/cAMP signalling interaction is implicated in the modulation of transmitters release from sympathetic neurons Caricati-Neto et al. [13], Bergantin & Caricati-Neto [14].
It is now well established that the Ca2+/cAMP signalling interaction is as a key cellular process in mammalians Caricati- Neto et al. [13], Bergantin & Caricati-Neto [14]. This nowadays accepted concept assumes that these signalling pathways virtually exist in all mammalian cells, regulated by adenylyl cyclases (ACs) and phosphodiesterases (PDEs) Caricati-Neto et al. [13], Bergantin & Caricati-Neto [14]. Indeed, endoplasmic reticulum (ER) Ca2+ channels, notable the Ca2+ channels regulated by ryanodine receptors (RyR), have particularly been a forefront for the Ca2+/ cAMP signalling interaction field. We recognized that Ca2+/cAMP signalling interaction rules an important participation in the regulation of transmitter release from neurons and neuroendocrine cells, and also neuronal death originated from neurodegenerative diseases Caricati-Neto et al. [13], Bergantin & Caricati-Neto [14]. Then, the interaction of Ca2+ and cAMP signalling pathways could be a new therapeutic goal for pharmaceuticals to treat neurodegenerative diseases like ALS.
Several medical reports have been proving that use of L-type CCBs in the antihypertensive therapy alleviates systemic arterial hypertension, but produces sympathetic hyperactivity Grossman & Messerli [22]. Despite these adverse effects of CCBs have been initially attributed to adjust reflex of arterial pressure, during almost four decades this enigmatic phenomenon named “calcium paradox” remained without additional explanation. Through an original experiment, we revealed that the increased transmitter release from sympathetic neurons induced by CCBs is due to its interference on the Ca2+/cAMP signalling interaction, thus resulting on “calcium paradox” phenomenon Bergantin et al. [23]. We confirmed that neurogenic contractions of the vas deferens were completely abolished by L-type CCBs in high concentrations (>1 μmol/L), but paradoxically increased in concentrations below 1 μmol/L, thus defined as sympathetic hyperactivity promoted by CCBs Kreye & Lut [18], French & Scott [19], Moritoki et al. [20]. Our studies clearly established that the paradoxical sympathetic hyperactivity is due to an augmentation of neurotransmitter release from sympathetic neurons achieved by L-type CCBs due to its interfering on the Ca2+/cAMP signalling interaction.
Indeed, many reports have shown that elevation of [cAMP] c reduces neuronal death triggered by cytosolic Ca2+ overload, stimulating neuroprotective response Sommer et al. [24], Xiao et al. [25]. As mentioned above, the L-type CCBs increase neurotransmitter release due to its interference on the Ca2+/cAMP signalling interaction. This interference results in the increase of ACs activity and elevation of [cAMP]c that, in turn, stimulates Ca2+ release from ER that increases neurotransmitter release Caricati- Neto et al. [13], Bergantin & Caricati-Neto [26]. In addition, this elevation of [cAMP]c produces neuroprotective response mediated by Ca2+/cAMP signalling interaction Caricati-Neto et al. [26]. Bergantin & Caricati-NetoIt [26], was proposed that this neuroprotective response results from activation by cAMP on the cellular survival pathways mediated by PKA/CREB Caricati-Neto et al. [26] Bergantin & Caricati-Neto [26]. Then, a new pharmacological goal for increasing neurotransmission in neurological and psychiatric disorders resulting of neurotransmitter release deficit, and neuronal death, could be achieved by the pharmacological interfering on the Ca2+/cAMP signalling interaction.
The combined use of the L-type CCBs, prescribed in the antihypertensive therapy, and cAMP-enhancer compounds, prescribed in the anti-depressive therapy such as rolipram, could be useful to achieve this purpose. It is important to note that the effect of this combined therapy in attenuating neuronal death may be related to the genomic response, as synaptic release may be attributed to a rapid response. Indeed, pharmacological modulation of the Ca2+/cAMP signaling interaction by combination of the L-type CCBs, and cAMP-enhancer compounds, could increase neurotransmission. In addition, pharmacological modulation of this interaction could subsidize the reducing of neuronal death due to attenuation of cytosolic Ca2+ overload, increase of [cAMP] c, and stimulation of cell survival pathways mediated by genomic response due to activation of cellular survival pathways regulated by cAMP/ PKA/CREB-dependent intracellular signaling pathway Ilijic et al. [21] , Hanon et al. [27], Onozuka et al. [28], (Figure 1) illustrates how to produce cellular responses: increase of neurotransmitter release (rapid response), and attenuation of neuronal death (genomic response), by the pharmacological modulation of the Ca2+/cAMP signalling interaction (Figure 1).
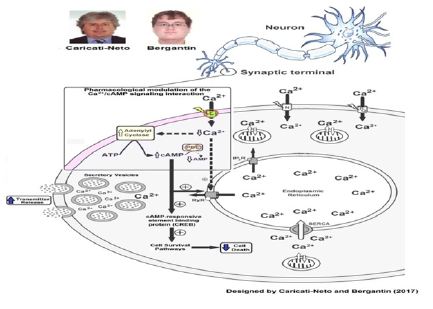
Figure 1: Pharmacological modulation of the Ca2+/cAMP signalling interaction as a new therapeutic strategy for ALS.
The reduction by CCBs of the Ca2+ influx through L-type voltageactivated Ca2+ channels enhances the adenylyl cyclase activity and consequently cAMP cytosolic levels. These CCBs-effects can be potentiated by cAMP-enhancer compounds (like PDEs inhibitors). Combined use of the CCBs with cAMP-enhancer compounds could attenuate the degeneration and death of the motor neurons triggered mainly by cytosolic Ca2+ overload (neuroprotector effect). Additionally, this pharmacological strategy could attenuate the deficit in the cholinergic neurotransmission in the neuromuscular synapses due to increase of acetylcholine release from motor neurons. Our studies suggest that this pharmacological strategy based in the modulation by drugs of the Ca2+/cAMP signalling interaction could be useful to attenuate the motor dysfunctions in patients with ALS Caricati-Neto et al. [13], & Bergantin , Caricati- Neto, [14].
In fact, it was demonstrated that the use of L-type CCBs reduces motor symptoms, and reduces progressive neuronal death in animal model of Parkinson´s disease, indicating that L-type CCBs are potentially viable neuroprotective pharmaceuticals Ilijic et al. [10]. Intriguingly, a 1-decade study involving thousands senile hypertensive patients demonstrated that prescription of L-type CCBs reduced blood pressure, and risk of dementia, in hypertensive patients, indicating that these pharmaceuticals could be clinically used to treat neurodegenerative diseases Wu & Wen [11]. These results for the neuroprotective effects of CCBs have been reinvestigated in thousands elderly hypertensive patients with memory dysfunction Hanon et al. [28]. In addition, these studies concluded that patients who have taken CCBs had their risk of cognitive dysfunction decreased, such as Alzheimer´s disease Hanon et al. [28]. Together, these findings reinforce the idea that reduction of cytosolic Ca2+ overload produced by L-type CCBs due to blockade of Ca2+ influx could be an alternative pharmacological goal to reduce, or prevent, neuronal death in neurodegenerative diseases like ALS. This neurological disease results from a neurodegenerative disorder that affects the motor control of skeletal muscles, producing the progressive loss of motor function [29,30]. It is caused by a loss, neuronal death, of specialized nerve cells, motor neurons. The loss of motor neurons leads to weakness and wasting, atrophy, of muscles used for activities such as crawling, walking, sitting up, and controlling of head movement. In severe cases of ALS, the muscles involved in breathing and swallowing are dramatically affected.
Based on previous described findings, we have anticipated a new therapeutic goal for increasing neurotransmission in neurological, and psychiatric disorders, resulting of neurotransmitter release deficit, and neuronal death originated from ALS Caricati-Neto et al. [13], Bergantin & Caricati-Neto, [14] the pharmacological regulation of the Ca2+/cAMP signalling interaction produced by combined use of the L-type CCBs and cAMP-enhancer compounds, which could open a new pathway to the drug development for the treatment of ALS and other neurodegenerative diseases Bergantin & Caricati-Neto[14].
In conclusion, pharmacological modulation of the Ca2+/ cAMP signalling interaction could be a more effective therapeutic approach for attenuating motor neuronal death and stimulating neuromuscular cholinergic neurotransmission compromised by acetylcholine release deficit in the ALS.


























